

Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies
- PMID: 26446103
- PMCID: PMC4698305
- DOI: 10.1007/s00401-015-1485-1
Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies
Abstract
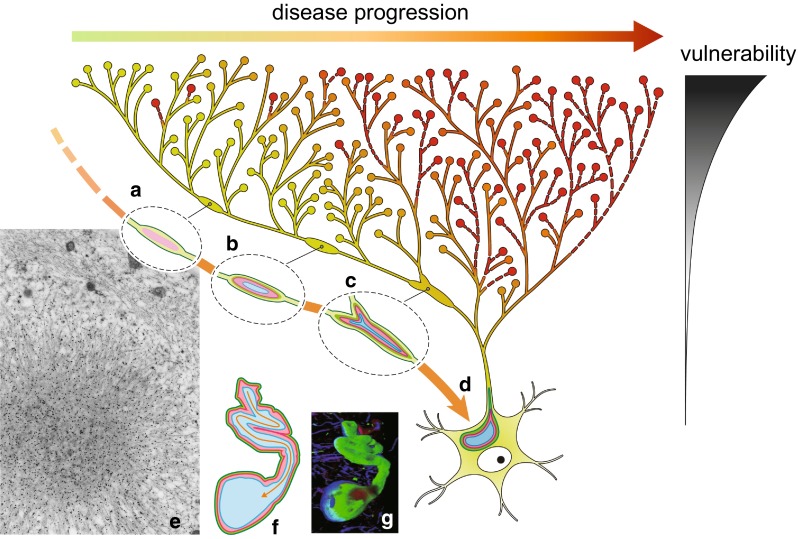
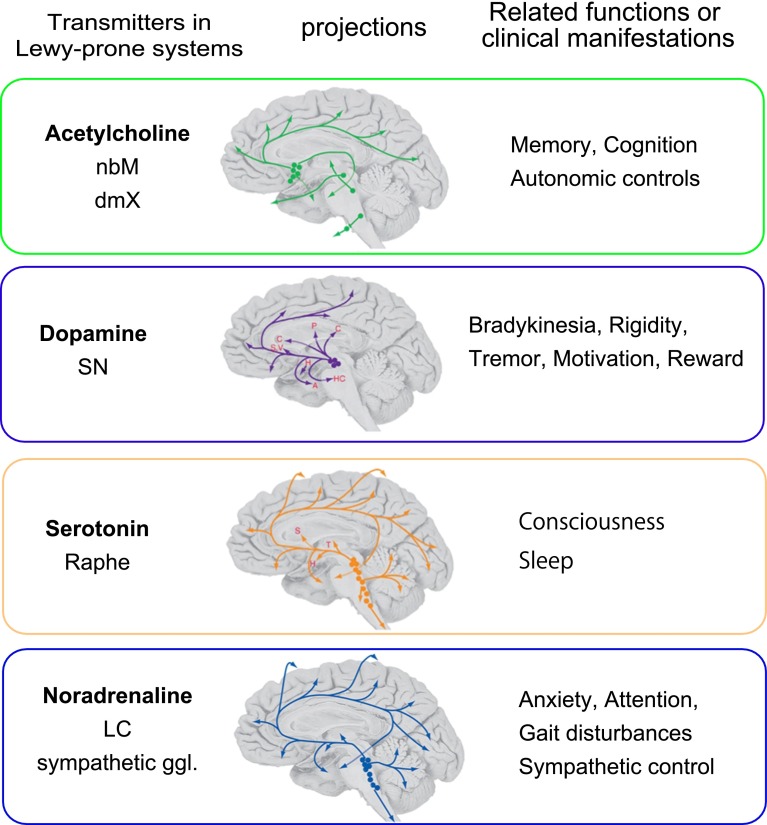
Progressive aggregation of alpha-synuclein (αS) through formation of amorphous pale bodies to mature Lewy bodies or in neuronal processes as Lewy neurites may be the consequence of conformational protein changes and accumulations, which structurally represents "molecular template". Focal initiation and subsequent spread along anatomically connected structures embody "structural template". To investigate the hypothesis that both processes might be closely associated and involved in the progression of αS pathology, which can be observed in human brains, αS amyloidogenic precursors termed "seeds" were experimentally injected into the brain or peripheral nervous system of animals. Although these studies showed that αS amyloidogenic seeds can induce αS pathology, which can spread in the nervous system, the findings are still not unequivocal in demonstrating predominant transsynaptic or intraneuronal spreads either in anterograde or retrograde directions. Interpretation of some of these studies is further complicated by other concurrent aberrant processes including neuroimmune activation, injury responses and/or general perturbation of proteostasis. In human brain, αS deposition and neuronal degeneration are accentuated in distal axon/synapse. Hyperbranching of axons is an anatomical commonality of Lewy-prone systems, providing a structural basis for abundance in distal axons and synaptic terminals. This neuroanatomical feature also can contribute to such distal accentuation of vulnerability in neuronal demise and the formation of αS inclusion pathology. Although retrograde progression of αS aggregation in hyperbranching axons may be a consistent feature of Lewy pathology, the regional distribution and gradient of Lewy pathology are not necessarily compatible with a predictable pattern such as upward progression from lower brainstem to cerebral cortex. Furthermore, "focal Lewy body disease" with the specific isolated involvement of autonomic, olfactory or cardiac systems suggests that spread of αS pathology is not always consistent. In many instances, the regional variability of Lewy pathology in human brain cannot be explained by a unified hypothesis such as transsynaptic spread. Thus, the distribution of Lewy pathology in human brain may be better explained by variable combinations of independent focal Lewy pathology to generate "multifocal Lewy body disease" that could be coupled with selective but variable neuroanatomical spread of αS pathology. More flexible models are warranted to take into account the relative propensity to develop Lewy pathology in different Lewy-prone systems, even without interconnections, compatible with the expanding clinicopathological spectra of Lewy-related disorders. These revised models are useful to better understand the mechanisms underlying the variable progression of Lewy body diseases so that diagnostic and therapeutic strategies are improved.
Keywords: Alpha-synuclein; Focal Lewy body disease; Gradient; Hyperbranching axons; Molecular template; Structural template.
Figures





References
-
- Alafuzoff I, Parkkinen L, Al-Sarraj S, Arzberger T, Bell J, Bodi I, Bogdanovic N, Budka H, Ferrer I, Gelpi E, Gentleman S, Giaccone G, Kamphorst W, King A, Korkolopoulou P, Kovacs GG, Larionov S, Meyronet D, Monoranu C, Morris J, Parchi P, Patsouris E, Roggendorf W, Seilhean D, Streichenberger N, Thal DR, Kretzschmar H, BrainNet Europe C. Assessment of alpha-synuclein pathology: a study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol. 2008;67(2):125–143. doi: 10.1097/nen.0b013e3181633526. - DOI - PubMed
-
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. Phosphorylation of ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281(40):29739–29752. doi: 10.1074/jbc.M600933200. - DOI - PubMed
-
- Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Jon SA, Farrer MJ. Alpha-synuclein p. H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord. 2013;28(6):811–813. doi: 10.1002/mds.25421. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
