

Coevolutionary Landscape Inference and the Context-Dependence of Mutations in Beta-Lactamase TEM-1
- PMID: 26446903
- PMCID: PMC4693977
- DOI: 10.1093/molbev/msv211
Coevolutionary Landscape Inference and the Context-Dependence of Mutations in Beta-Lactamase TEM-1
Abstract
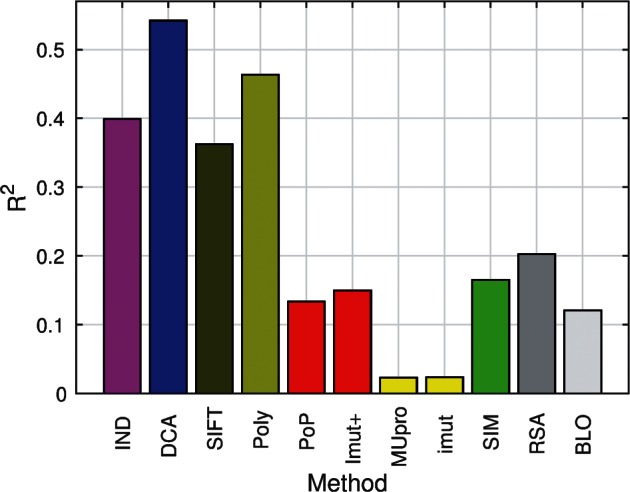
The quantitative characterization of mutational landscapes is a task of outstanding importance in evolutionary and medical biology: It is, for example, of central importance for our understanding of the phenotypic effect of mutations related to disease and antibiotic drug resistance. Here we develop a novel inference scheme for mutational landscapes, which is based on the statistical analysis of large alignments of homologs of the protein of interest. Our method is able to capture epistatic couplings between residues, and therefore to assess the dependence of mutational effects on the sequence context where they appear. Compared with recent large-scale mutagenesis data of the beta-lactamase TEM-1, a protein providing resistance against beta-lactam antibiotics, our method leads to an increase of about 40% in explicative power as compared with approaches neglecting epistasis. We find that the informative sequence context extends to residues at native distances of about 20 Å from the mutated site, reaching thus far beyond residues in direct physical contact.
Keywords: coevolution; epistasis; genotype–phenotype mapping; mutational landscape; statistical inference.
© The Author 2015. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
