

Metabolic and Phenotypic Differences between Mice Producing a Werner Syndrome Helicase Mutant Protein and Wrn Null Mice
- PMID: 26447695
- PMCID: PMC4598085
- DOI: 10.1371/journal.pone.0140292
Metabolic and Phenotypic Differences between Mice Producing a Werner Syndrome Helicase Mutant Protein and Wrn Null Mice
Abstract
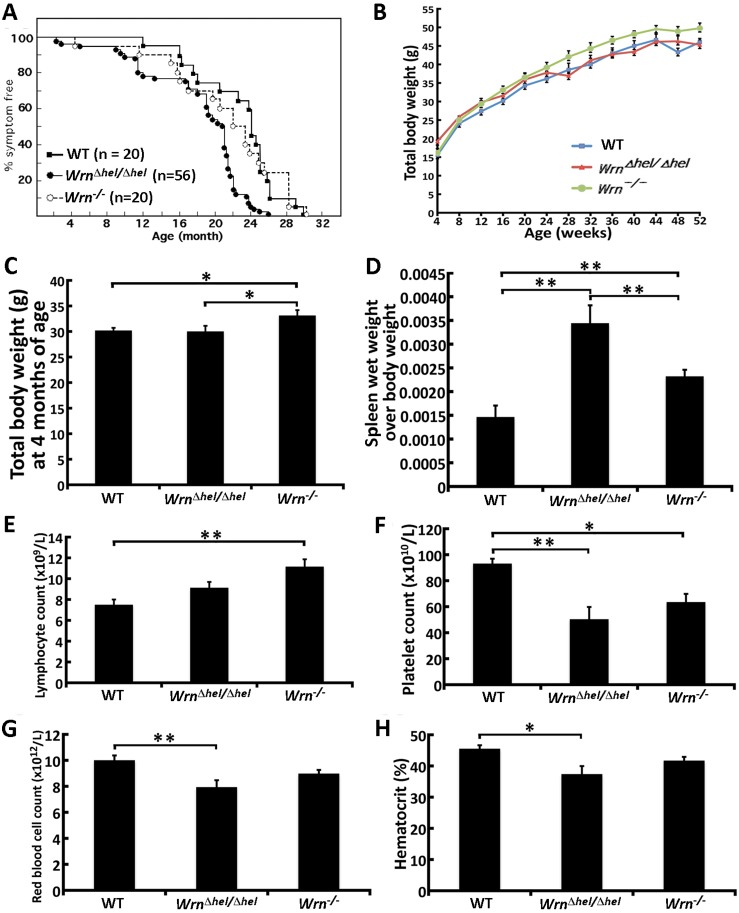
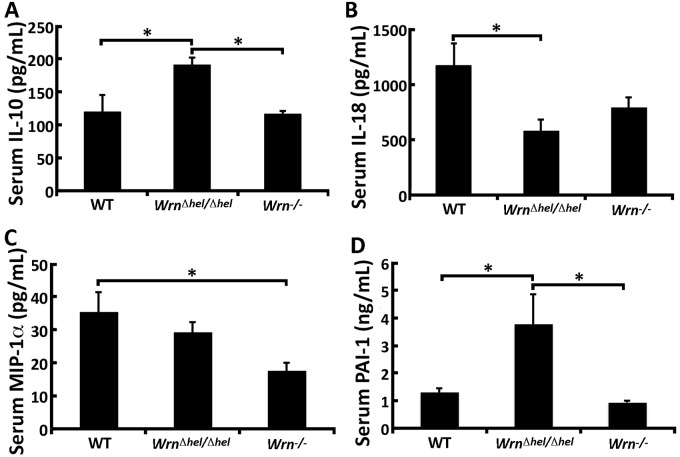
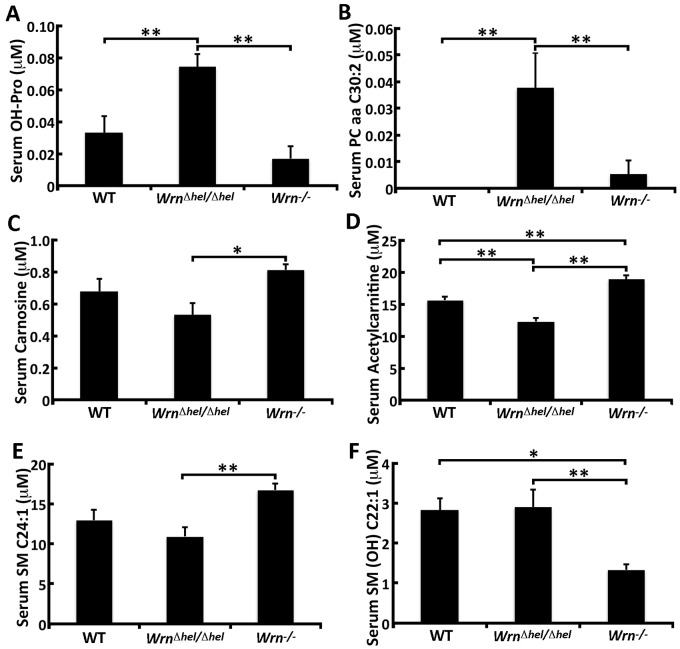
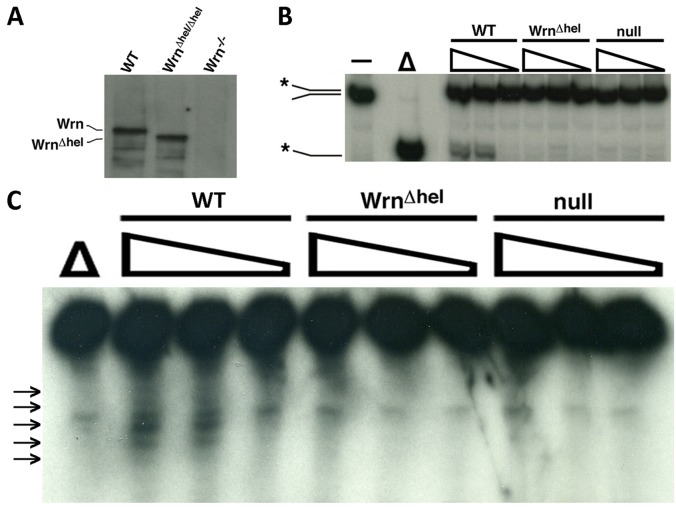
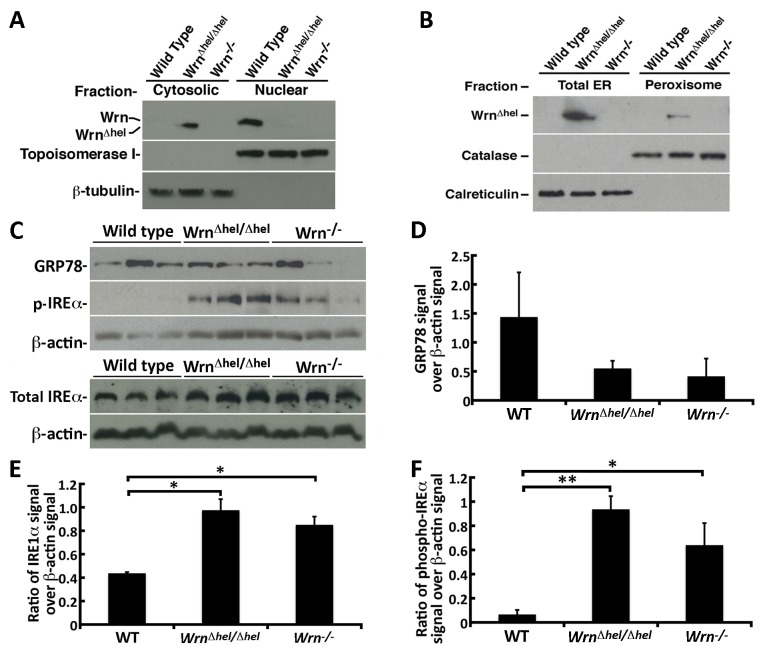
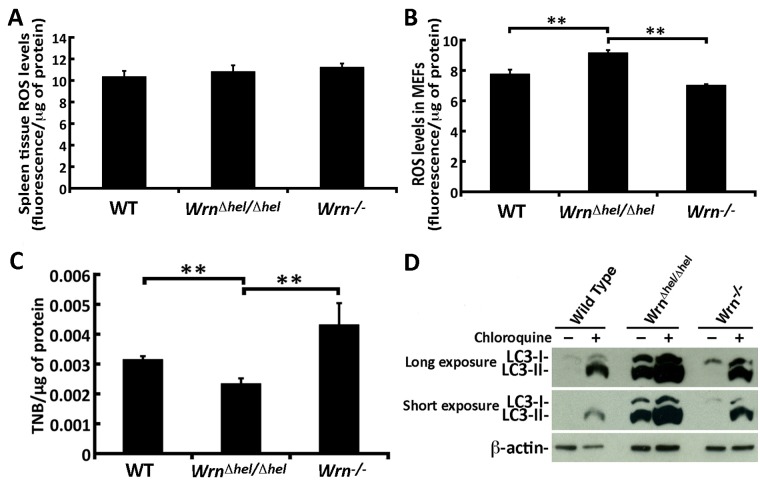
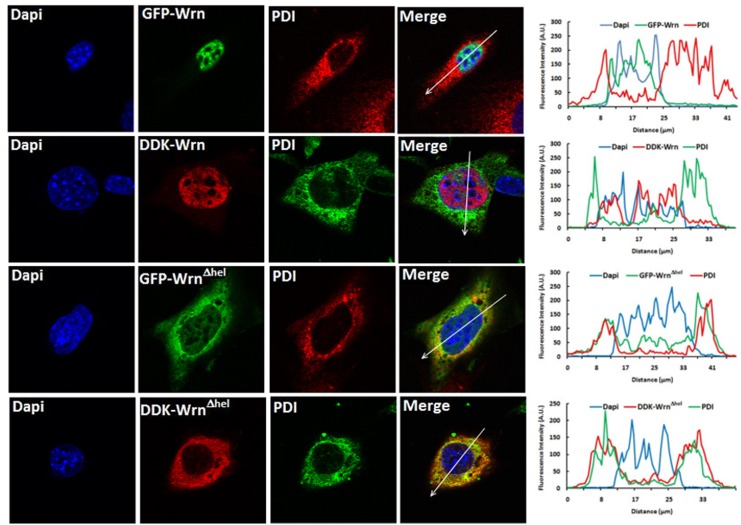
Werner syndrome (WS) is a premature aging disorder caused by mutations in a RecQ-family DNA helicase, WRN. Mice lacking part of the helicase domain of the WRN orthologue exhibit many phenotypic features of WS, including metabolic abnormalities and a shorter mean life span. In contrast, mice lacking the entire Wrn protein (i.e. Wrn null mice) do not exhibit a premature aging phenotype. In this study, we used a targeted mass spectrometry-based metabolomic approach to identify serum metabolites that are differentially altered in young Wrn helicase mutant and Wrn null mice. An antibody-based quantification of 43 serum cytokines and markers of cardiovascular disease risk complemented this study. We found that Wrn helicase mutants exhibited elevated and decreased levels, respectively, of the anti-inflammatory cytokine IL-10 and the pro-inflammatory cytokine IL-18. Wrn helicase mutants also exhibited an increase in serum hydroxyproline and plasminogen activator inhibitor-1, markers of extracellular matrix remodeling of the vascular system and inflammation in aging. We also observed an abnormal increase in the ratio of very long chain to short chain lysophosphatidylcholines in the Wrn helicase mutants underlying a peroxisome perturbation in these mice. Remarkably, the Wrn mutant helicase protein was mislocalized to the endoplasmic reticulum and the peroxisomal fractions in liver tissues. Additional analyses with mouse embryonic fibroblasts indicated a severe defect of the autophagy flux in cells derived from Wrn helicase mutants compared to wild type and Wrn null animals. These results indicate that the deleterious effects of the helicase-deficient Wrn protein are mediated by the dysfunction of several cellular organelles.
Conflict of interest statement
Figures
References
-
- Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner's syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore). 1966;45(3):177–221. Epub 1966/05/01. . - PubMed
-
- Salk D. Werner's syndrome: a review of recent research with an analysis of connective tissue metabolism, growth control of cultured cells, and chromosomal aberrations. Hum Genet. 1982;62(1):1–5. Epub 1982/01/01. . - PubMed
-
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, et al. Positional cloning of the Werner's syndrome gene. Science. 1996;272(5259):258–62. Epub 1996/04/12. . - PubMed
-
- Kamath-Loeb AS, Shen JC, Loeb LA, Fry M. Werner syndrome protein. II. Characterization of the integral 3'—> 5' DNA exonuclease. J Biol Chem. 1998;273(51):34145–50. Epub 1998/12/16. . - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
