

A maximum pseudo-likelihood approach for phylogenetic networks
- PMID: 26450642
- PMCID: PMC4602316
- DOI: 10.1186/1471-2164-16-S10-S10
A maximum pseudo-likelihood approach for phylogenetic networks
Abstract
Background: Several phylogenomic analyses have recently demonstrated the need to account simultaneously for incomplete lineage sorting (ILS) and hybridization when inferring a species phylogeny. A maximum likelihood approach was introduced recently for inferring species phylogenies in the presence of both processes, and showed very good results. However, computing the likelihood of a model in this case is computationally infeasible except for very small data sets.
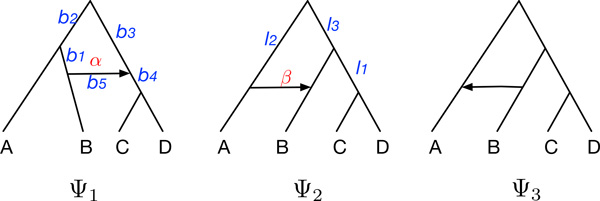
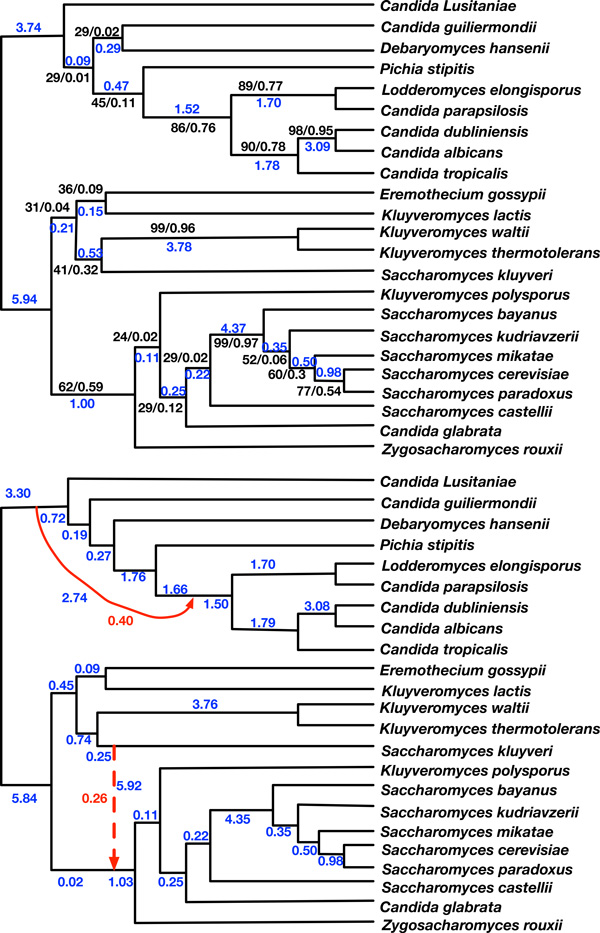
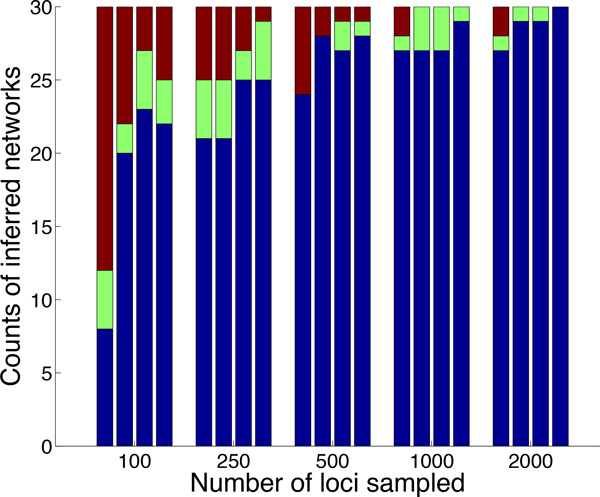
Results: Inspired by recent work on the pseudo-likelihood of species trees based on rooted triples, we introduce the pseudo-likelihood of a phylogenetic network, which, when combined with a search heuristic, provides a statistical method for phylogenetic network inference in the presence of ILS. Unlike trees, networks are not always uniquely encoded by a set of rooted triples. Therefore, even when given sufficient data, the method might converge to a network that is equivalent under rooted triples to the true one, but not the true one itself. The method is computationally efficient and has produced very good results on the data sets we analyzed. The method is implemented in PhyloNet, which is publicly available in open source.
Conclusions: Maximum pseudo-likelihood allows for inferring species phylogenies in the presence of hybridization and ILS, while scaling to much larger data sets than is currently feasible under full maximum likelihood. The nonuniqueness of phylogenetic networks encoded by a system of rooted triples notwithstanding, the proposed method infers the correct network under certain scenarios, and provides candidates for further exploration under other criteria and/or data in other scenarios.
Figures
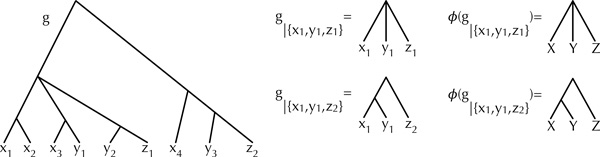
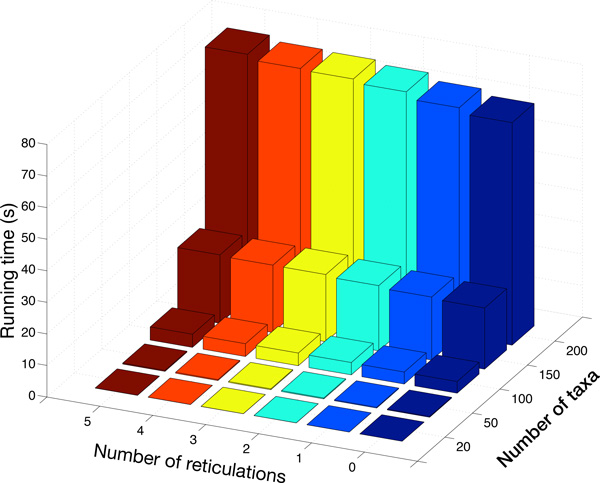
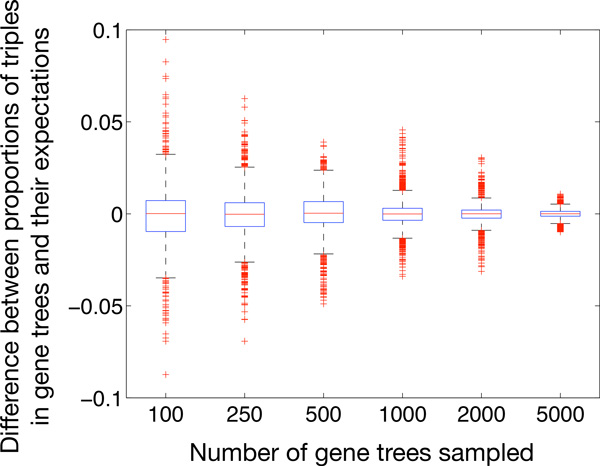
References
-
- Arnold ML. Natural Hybridization and Evolution. Oxford University Press, Oxford; 1997.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
