

Ancestral gene synteny reconstruction improves extant species scaffolding
- PMID: 26450761
- PMCID: PMC4603332
- DOI: 10.1186/1471-2164-16-S10-S11
Ancestral gene synteny reconstruction improves extant species scaffolding
Abstract
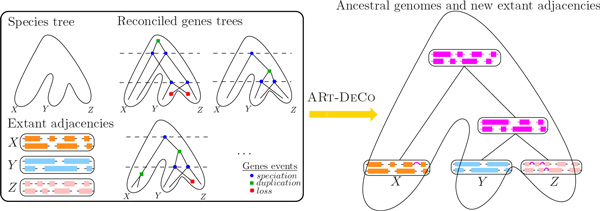
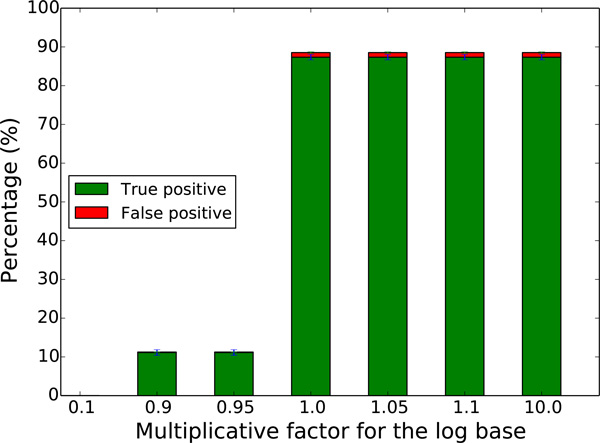
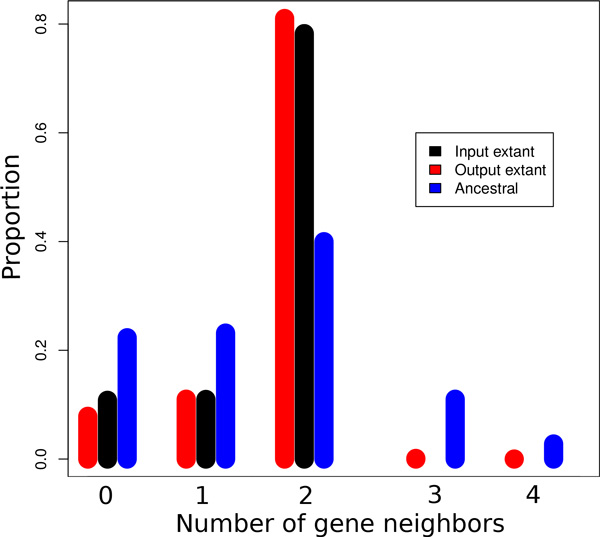
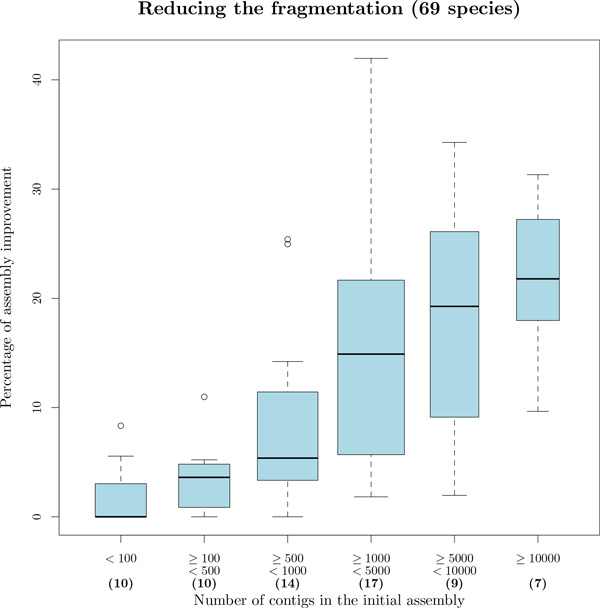
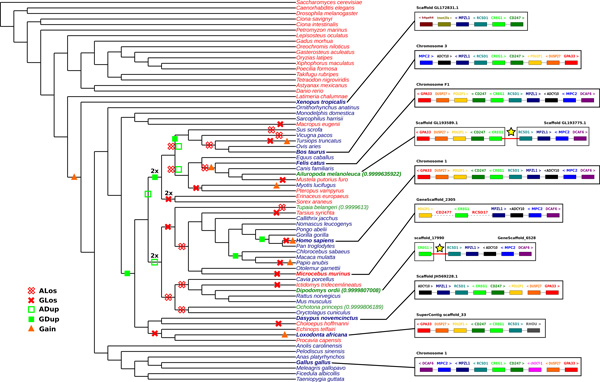
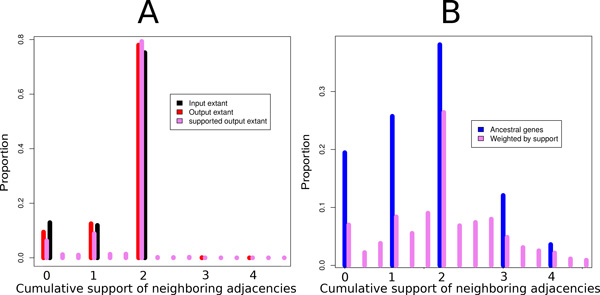
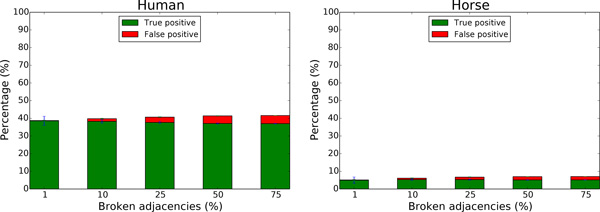
We exploit the methodological similarity between ancestral genome reconstruction and extant genome scaffolding. We present a method, called ARt-DeCo that constructs neighborhood relationships between genes or contigs, in both ancestral and extant genomes, in a phylogenetic context. It is able to handle dozens of complete genomes, including genes with complex histories, by using gene phylogenies reconciled with a species tree, that is, annotated with speciation, duplication and loss events. Reconstructed ancestral or extant synteny comes with a support computed from an exhaustive exploration of the solution space. We compare our method with a previously published one that follows the same goal on a small number of genomes with universal unicopy genes. Then we test it on the whole Ensembl database, by proposing partial ancestral genome structures, as well as a more complete scaffolding for many partially assembled genomes on 69 eukaryote species. We carefully analyze a couple of extant adjacencies proposed by our method, and show that they are indeed real links in the extant genomes, that were missing in the current assembly. On a reduced data set of 39 eutherian mammals, we estimate the precision and sensitivity of ARt-DeCo by simulating a fragmentation in some well assembled genomes, and measure how many adjacencies are recovered. We find a very high precision, while the sensitivity depends on the quality of the data and on the proximity of closely related genomes.
Figures
References
-
- Raphael BJ, Volik S, Collins C, Pevzner PA. Reconstructing tumor genome architectures. Bioinformatics. 2003;19(Suppl. 2) - PubMed
-
- McPherson A, Roth A, Ha G, Shah SP, Chauve C, Sahinalp SC. Joint inference of genome structure and content in heterogeneous tumor samples. Research in Computational Molecular Biology Lecture Notes in Computer Science. 2015;9029:256–258.
-
- Hurst L, Pál C, Lercher M. The evolutionary dynamics of eukaryotic gene order. Nat Rev Genet. 2004;5(4):299–310. - PubMed
-
- Swenson K, Arndt W, Tang J, Moret B. Phylogenetic reconstruction from complete gene orders of whole genomes. Proceedings of the 6th Asia Pacific Bioinformatics Conference. 2008. pp. 241–250.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
