

Refining Genotype-Phenotype Correlation in Autosomal Dominant Polycystic Kidney Disease
- PMID: 26453610
- PMCID: PMC4884120
- DOI: 10.1681/ASN.2015060648
Refining Genotype-Phenotype Correlation in Autosomal Dominant Polycystic Kidney Disease
Abstract
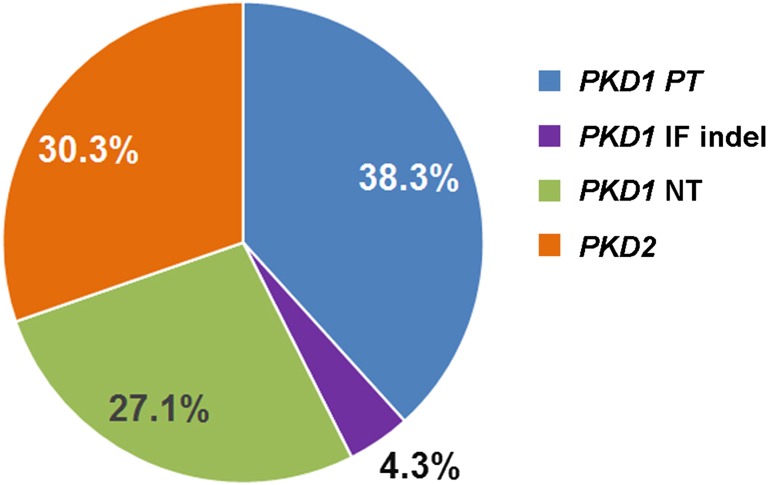
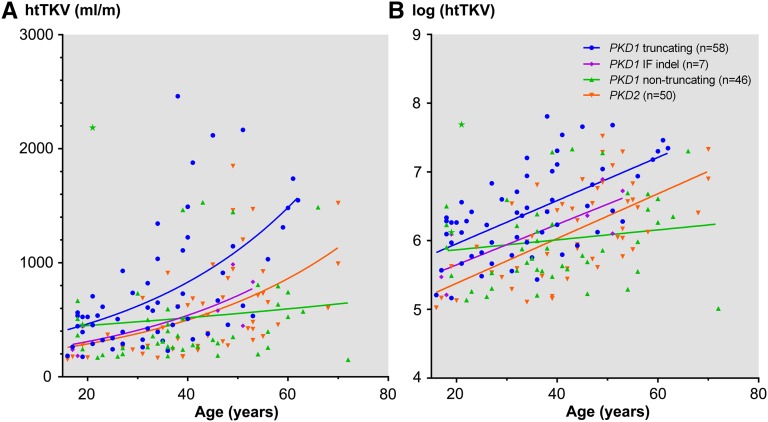
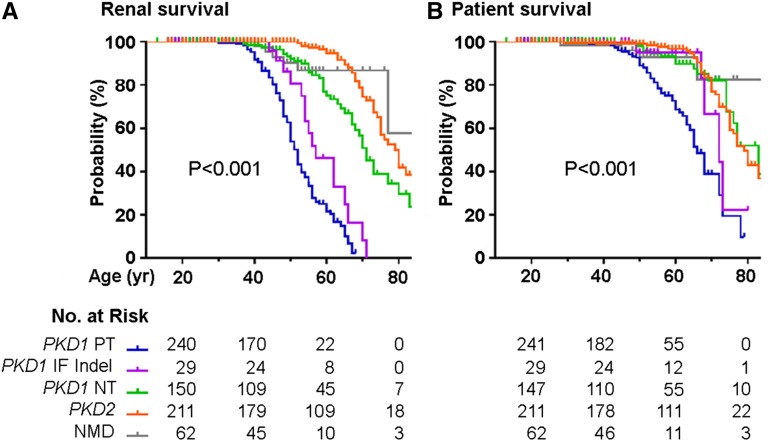
Renal disease variability in autosomal dominant polycystic kidney disease (ADPKD) is strongly influenced by the gene locus (PKD1 versus PKD2). Recent studies identified nontruncating PKD1 mutations in approximately 30% of patients who underwent comprehensive mutation screening, but the clinical significance of these mutations is not well defined. We examined the genotype-renal function correlation in a prospective cohort of 220 unrelated ADPKD families ascertained through probands with serum creatinine ≤1.4 mg/dl at recruitment. We screened these families for PKD1 and PKD2 mutations and reviewed the clinical outcomes of the probands and affected family members. Height-adjusted total kidney volume (htTKV) was obtained in 161 affected subjects. Multivariate Cox proportional hazard modeling for renal and patient survival was performed in 707 affected probands and family members. Overall, we identified pathogenic mutations in 84.5% of our families, in which the prevalence of PKD1 truncating, PKD1 in-frame insertion/deletion, PKD1 nontruncating, and PKD2 mutations was 38.3%, 4.3%, 27.1%, and 30.3%, respectively. Compared with patients with PKD1 truncating mutations, patients with PKD1 in-frame insertion/deletion, PKD1 nontruncating, or PKD2 mutations have smaller htTKV and reduced risks (hazard ratio [95% confidence interval]) of ESRD (0.35 [0.14 to 0.91], 0.10 [0.05 to 0.18], and 0.03 [0.01 to 0.05], respectively) and death (0.31 [0.11 to 0.87], 0.20 [0.11 to 0.38], and 0.18 [0.11 to 0.31], respectively). Refined genotype-renal disease correlation coupled with targeted next generation sequencing of PKD1 and PKD2 may provide useful clinical prognostication for ADPKD.
Keywords: ADPKD; genetic renal disease; genetics and development.
Copyright © 2016 by the American Society of Nephrology.
Figures
References
-
- Spithoven EM, Kramer A, Meijer E, Orskov B, Wanner C, Abad JM, Aresté N, de la Torre RA, Caskey F, Couchoud C, Finne P, Heaf J, Hoitsma A, de Meester J, Pascual J, Postorino M, Ravani P, Zurriaga O, Jager KJ, Gansevoort RT ERA-EDTA Registry EuroCYST Consortium WGIKD : Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: Prevalence and survival—an analysis of data from the ERA-EDTA Registry. Nephrol Dial Transplant 29[Suppl 4]: iv15–iv25, 2014 - PMC - PubMed
-
- Peters DJ, Sandkuijl LA: Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol 97: 128–139, 1992 - PubMed
-
- Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D: Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet 353: 103–107, 1999 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
