

Bayesian integration of genetics and epigenetics detects causal regulatory SNPs underlying expression variability
- PMID: 26456756
- PMCID: PMC4633824
- DOI: 10.1038/ncomms9555
Bayesian integration of genetics and epigenetics detects causal regulatory SNPs underlying expression variability
Abstract
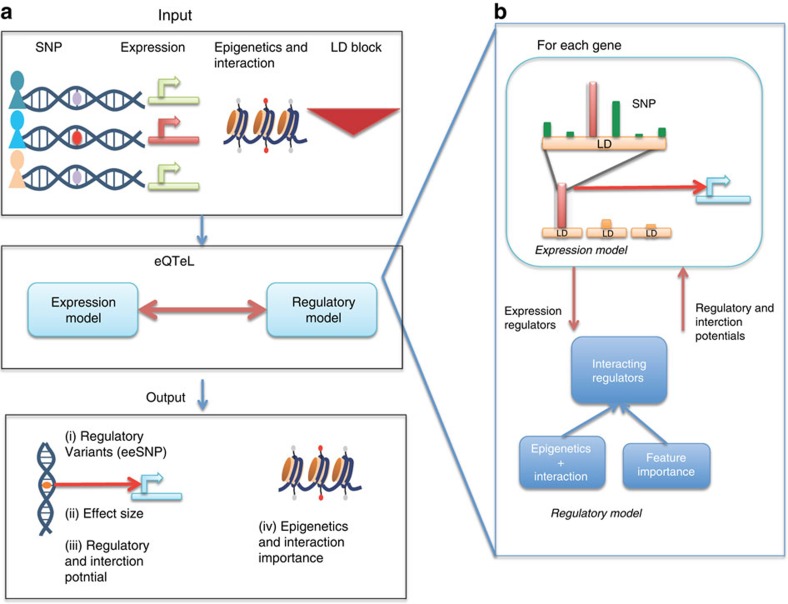
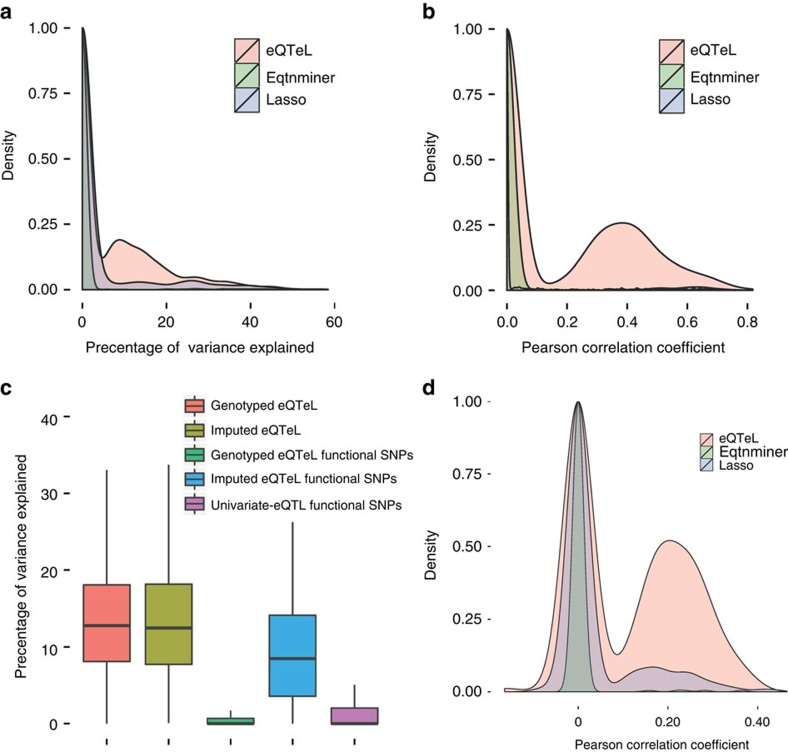
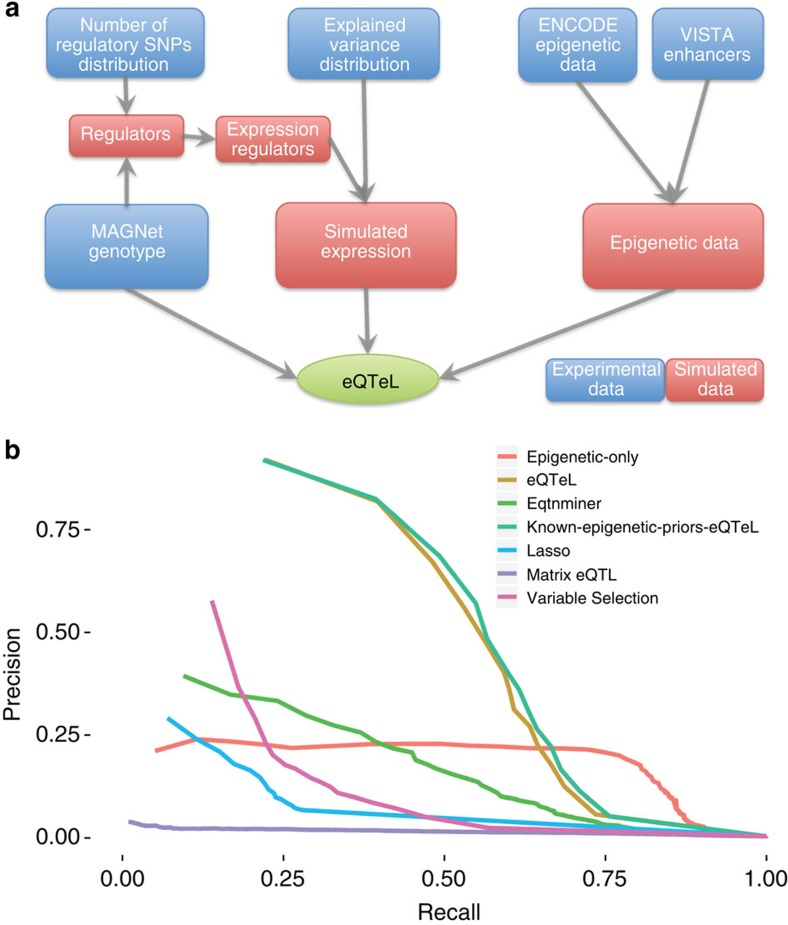
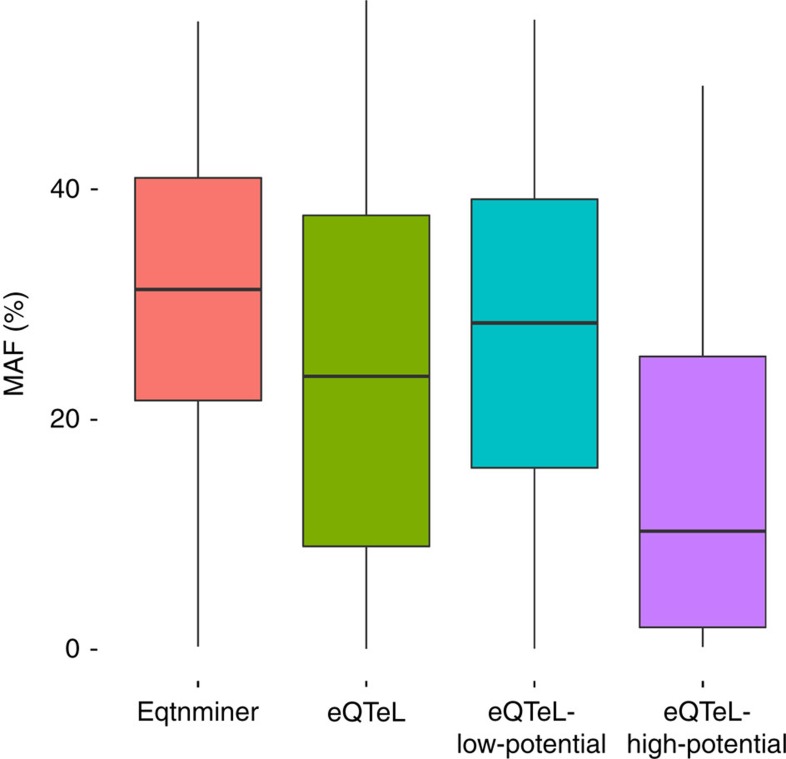
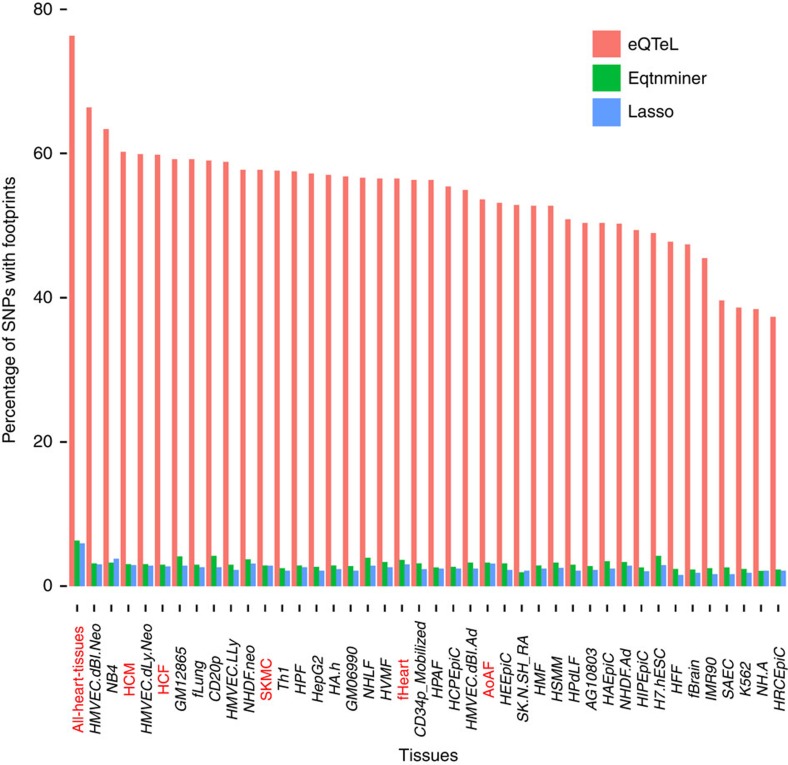
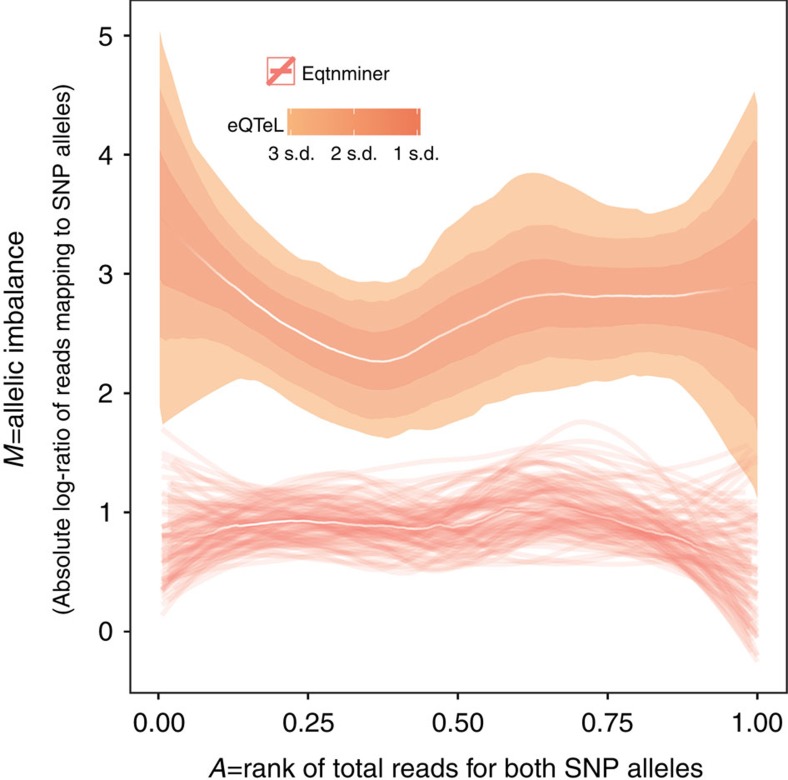
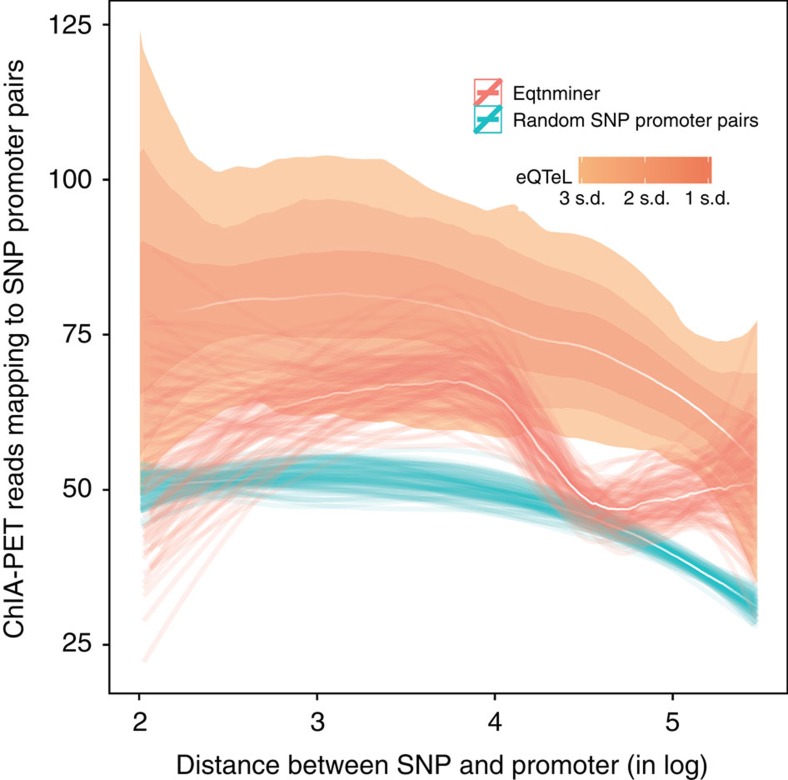
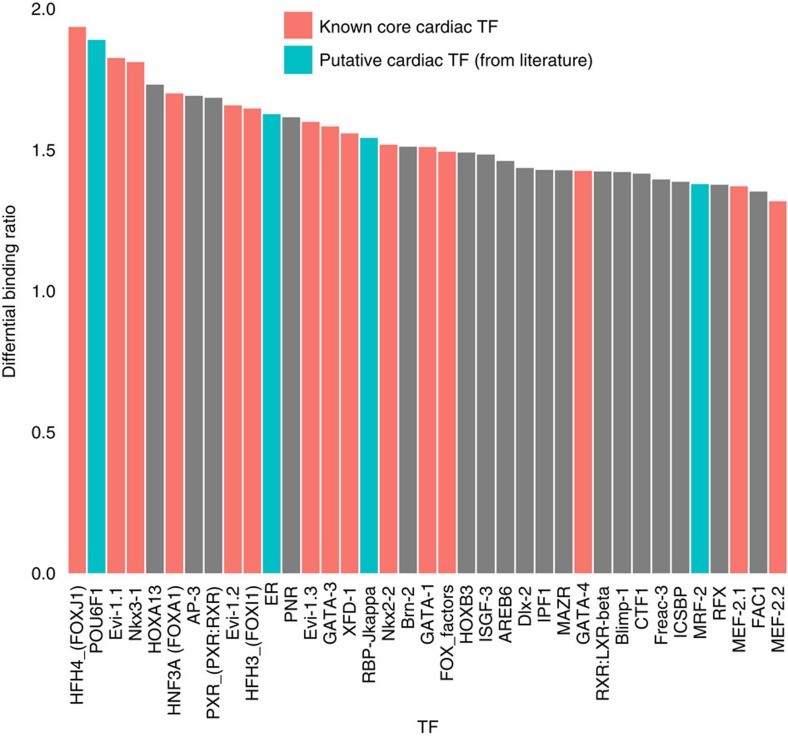
The standard expression quantitative trait loci (eQTL) detects polymorphisms associated with gene expression without revealing causality. We introduce a coupled Bayesian regression approach--eQTeL, which leverages epigenetic data to estimate regulatory and gene interaction potential, and identifies combination of regulatory single-nucleotide polymorphisms (SNPs) that explain the gene expression variance. On human heart data, eQTeL not only explains a significantly greater proportion of expression variance but also predicts gene expression more accurately than other methods. Based on realistic simulated data, we demonstrate that eQTeL accurately detects causal regulatory SNPs, including those with small effect sizes. Using various functional data, we show that SNPs detected by eQTeL are enriched for allele-specific protein binding and histone modifications, which potentially disrupt binding of core cardiac transcription factors and are spatially proximal to their target. eQTeL SNPs capture a substantial proportion of genetic determinants of expression variance and we estimate that 58% of these SNPs are putatively causal.
Figures
References
-
- Beyer K. & Goldstein J. When is nearest neighbour meaningful? Database TheoryICDT'99 (1999). URL http://link.springer.com/chapter/10.1007/3-540-49257-7/_15. - DOI
-
- Kraft P. & Hunter D. Genetic risk prediction: are we there yet? N. Engl. J. Med. 360, 1701–1703 (2009). - PubMed
-
- Hirschhorn J. N. Genomewide association studies-illuminating biologic pathways. N. Engl. J. Med. 360, 1699–1701 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
