

Instability in progressive multiple sequence alignment algorithms
- PMID: 26457114
- PMCID: PMC4599319
- DOI: 10.1186/s13015-015-0057-1
Instability in progressive multiple sequence alignment algorithms
Abstract
Background: Progressive alignment is the standard approach used to align large numbers of sequences. As with all heuristics, this involves a tradeoff between alignment accuracy and computation time.
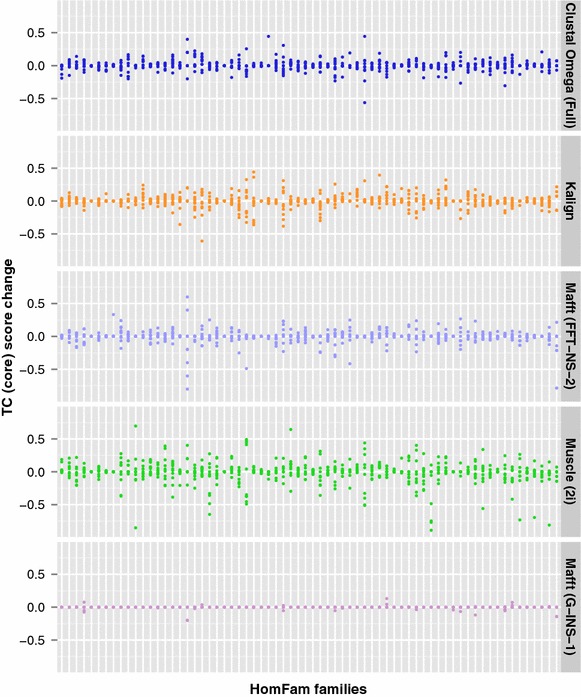
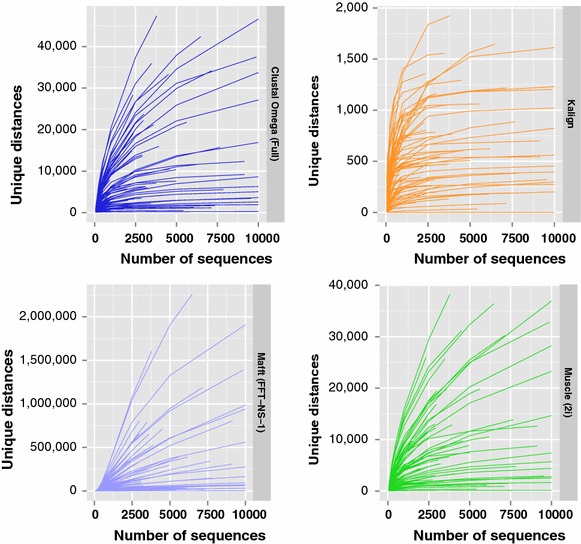
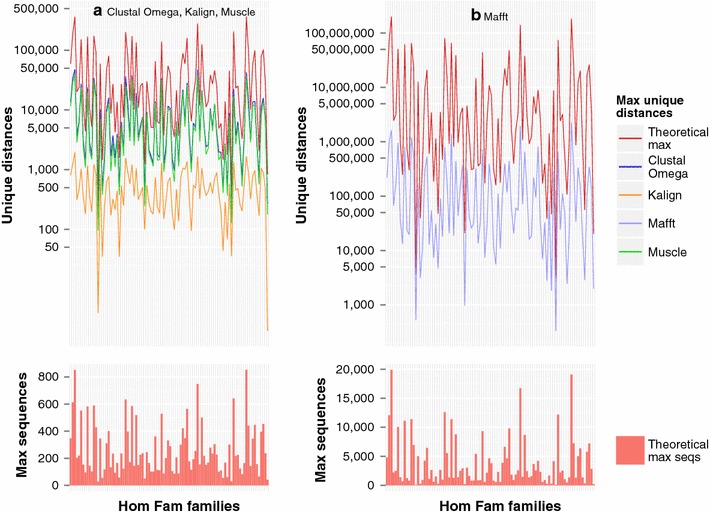
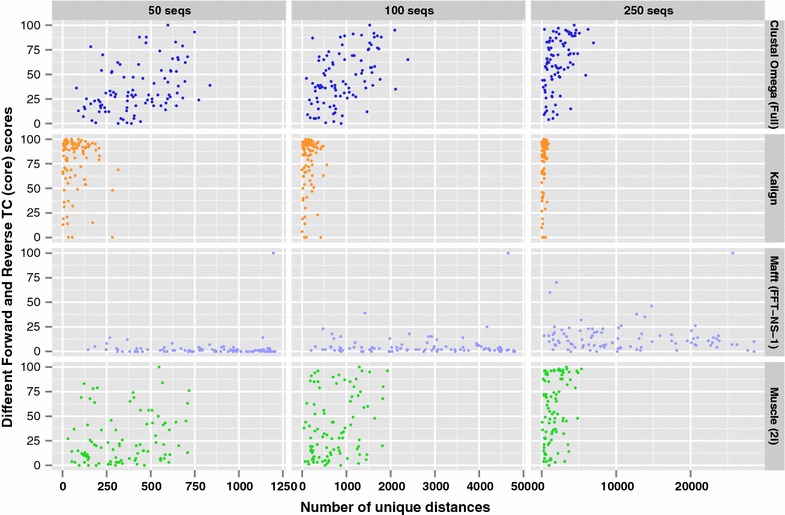
Results: We examine this tradeoff and find that, because of a loss of information in the early steps of the approach, the alignments generated by the most common multiple sequence alignment programs are inherently unstable, and simply reversing the order of the sequences in the input file will cause a different alignment to be generated. Although this effect is more obvious with larger numbers of sequences, it can also be seen with data sets in the order of one hundred sequences. We also outline the means to determine the number of sequences in a data set beyond which the probability of instability will become more pronounced.
Conclusions: This has major ramifications for both the designers of large-scale multiple sequence alignment algorithms, and for the users of these alignments.
Keywords: Clustal; Kalign; Large scale alignment; Mafft; Multiple sequence alignment; Muscle; Pfam; Sequence order.
Figures
References
-
- Higgins DG, Bleasby AJ, Fuchs R. CLUSTAL V: improved software for multiple sequence alignment. Comp Appl Biosci CABIOS. 1992;8(2):189–191. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
