

Estimating Metabolic Fluxes Using a Maximum Network Flexibility Paradigm
- PMID: 26457579
- PMCID: PMC4601694
- DOI: 10.1371/journal.pone.0139665
Estimating Metabolic Fluxes Using a Maximum Network Flexibility Paradigm
Abstract
Motivation: Genome-scale metabolic networks can be modeled in a constraint-based fashion. Reaction stoichiometry combined with flux capacity constraints determine the space of allowable reaction rates. This space is often large and a central challenge in metabolic modeling is finding the biologically most relevant flux distributions. A widely used method is flux balance analysis (FBA), which optimizes a biologically relevant objective such as growth or ATP production. Although FBA has proven to be highly useful for predicting growth and byproduct secretion, it cannot predict the intracellular fluxes under all environmental conditions. Therefore, alternative strategies have been developed to select flux distributions that are in agreement with experimental "omics" data, or by incorporating experimental flux measurements. The latter, unfortunately can only be applied to a limited set of reactions and is currently not feasible at the genome-scale. On the other hand, it has been observed that micro-organisms favor a suboptimal growth rate, possibly in exchange for a more "flexible" metabolic network. Instead of dedicating the internal network state to an optimal growth rate in one condition, a suboptimal growth rate is used, that allows for an easier switch to other nutrient sources. A small decrease in growth rate is exchanged for a relatively large gain in metabolic capability to adapt to changing environmental conditions.
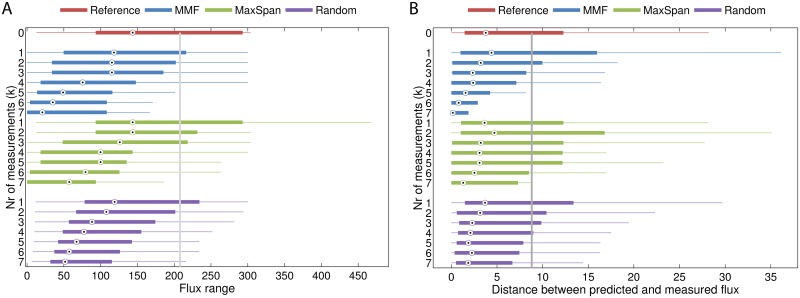
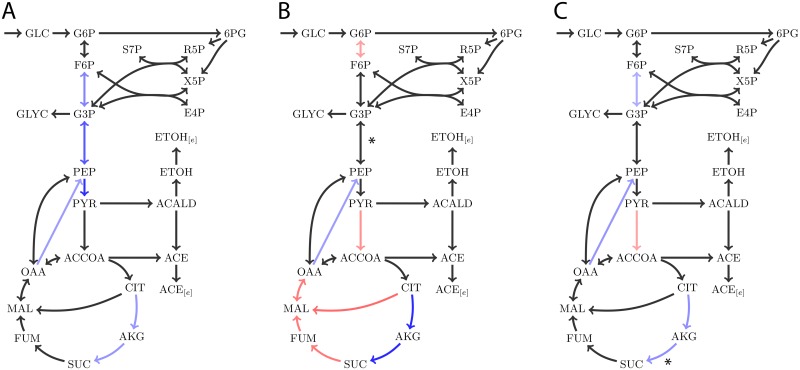
Results: Here, we propose Maximum Metabolic Flexibility (MMF) a computational method that utilizes this observation to find the most probable intracellular flux distributions. By mapping measured flux data from central metabolism to the genome-scale models of Escherichia coli and Saccharomyces cerevisiae we show that i) indeed, most of the measured fluxes agree with a high adaptability of the network, ii) this result can be used to further reduce the space of feasible solutions iii) this reduced space improves the quantitative predictions made by FBA and contains a significantly larger fraction of the measured fluxes compared to the flux space that was reduced by a uniform sampling approach and iv) MMF can be used to select reactions in the network that contribute most to the steady-state flux space. Constraining the selected reactions improves the quantitative predictions of FBA considerably more than adding an equal amount of flux constraints, selected using a more naïve approach. Our method can be applied to any cell type without requiring prior information.
Availability: MMF is freely available as a MATLAB plugin at: http://cs.ru.nl/~wmegchel/mmf.
Conflict of interest statement
Figures
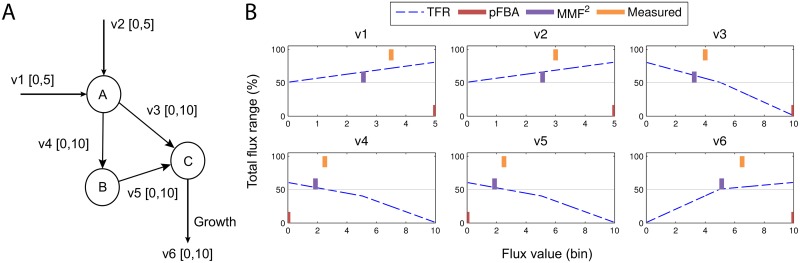
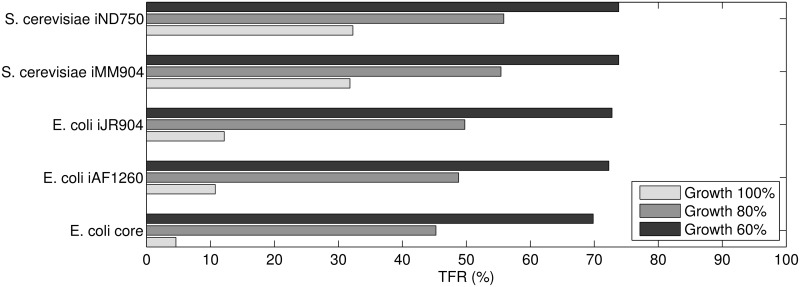
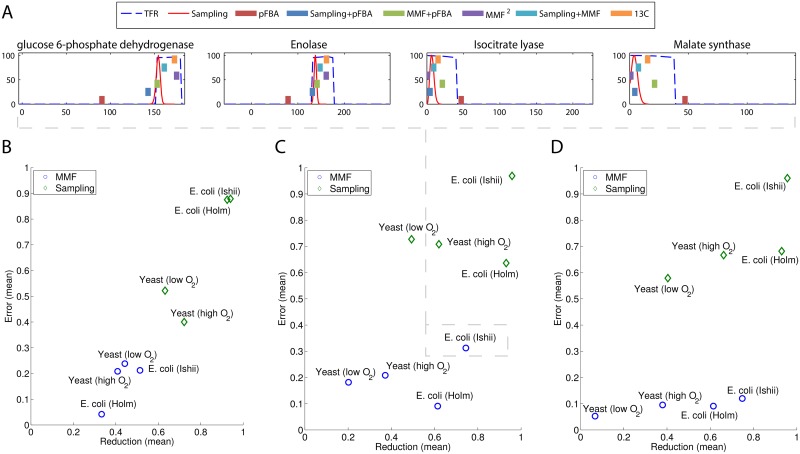
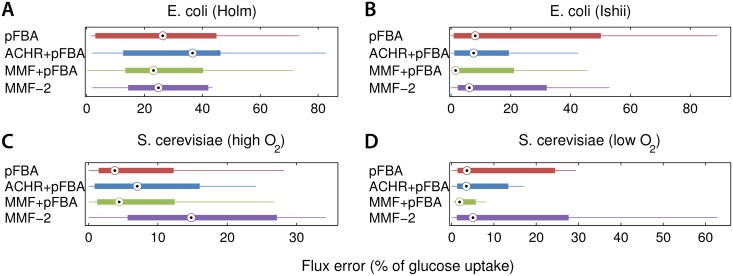
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
