

Duchenne Muscular Dystrophy: From Diagnosis to Therapy
- PMID: 26457695
- PMCID: PMC6332113
- DOI: 10.3390/molecules201018168
Duchenne Muscular Dystrophy: From Diagnosis to Therapy
Abstract
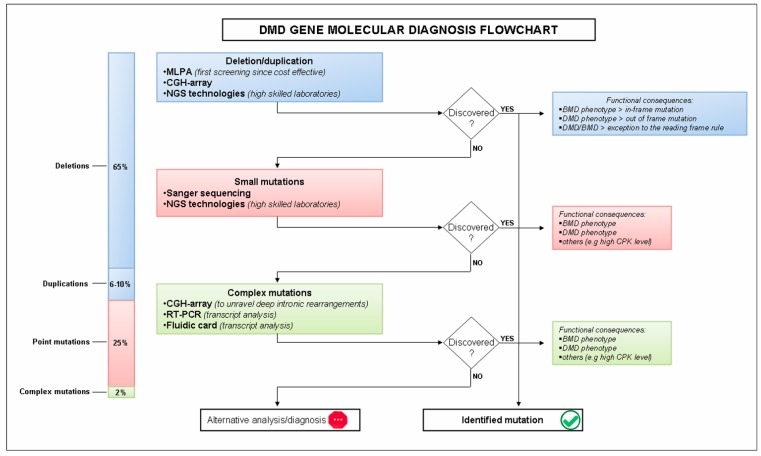
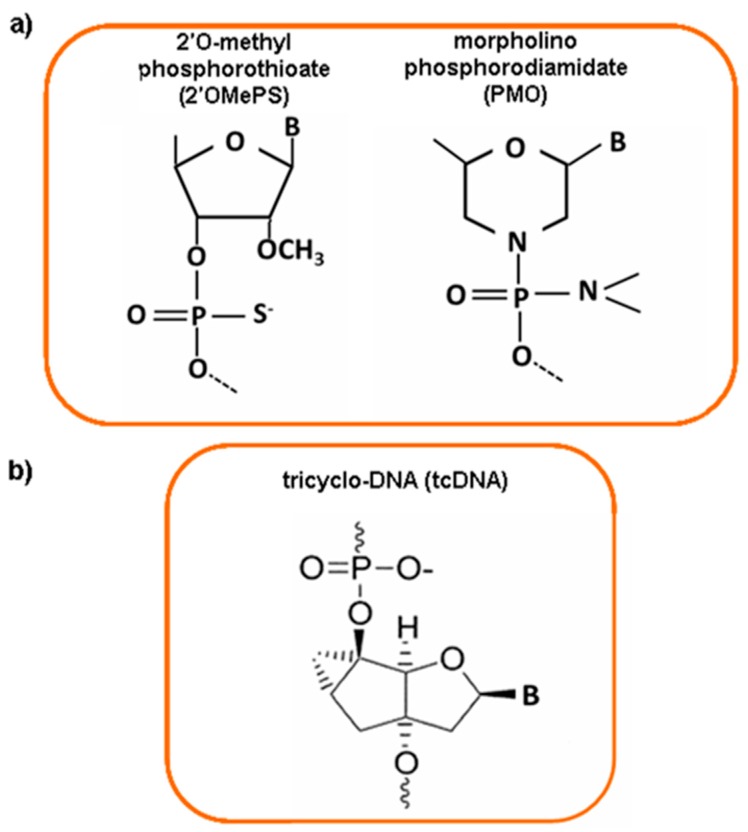
Duchenne muscular dystrophy (DMD) is an X-linked inherited neuromuscular disorder due to mutations in the dystrophin gene. It is characterized by progressive muscle weakness and wasting due to the absence of dystrophin protein that causes degeneration of skeletal and cardiac muscle. The molecular diagnostic of DMD involves a deletions/duplications analysis performed by quantitative technique such as microarray-based comparative genomic hybridization (array-CGH), Multiple Ligation Probe Assay MLPA. Since traditional methods for detection of point mutations and other sequence variants require high cost and are time consuming, especially for a large gene like dystrophin, the use of next-generation sequencing (NGS) has become a useful tool available for clinical diagnosis. The dystrophin gene is large and finely regulated in terms of tissue expression, and RNA processing and editing includes a variety of fine tuned processes. At present, there are no effective treatments and the steroids are the only fully approved drugs used in DMD therapy able to slow disease progression. In the last years, an increasing variety of strategies have been studied as a possible therapeutic approach aimed to restore dystrophin production and to preserve muscle mass, ameliorating the DMD phenotype. RNA is the most studied target for the development of clinical strategies and Antisense Oligonucleotides (AONs) are the most used molecules for RNA modulation. The identification of delivery system to enhance the efficacy and to reduce the toxicity of AON is the main purpose in this area and nanomaterials are a very promising model as DNA/RNA molecules vectors. Dystrophinopathies therefore represent a pivotal field of investigation, which has opened novel avenues in molecular biology, medical genetics and novel therapeutic options.
Keywords: Antisense Oligonucleotides; DMD therapy; antisense delivery; dystrophin; molecular diagnosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
