

Uncovering the Mechanism of Aggregation of Human Transthyretin
- PMID: 26459562
- PMCID: PMC4661406
- DOI: 10.1074/jbc.M115.659912
Uncovering the Mechanism of Aggregation of Human Transthyretin
Abstract
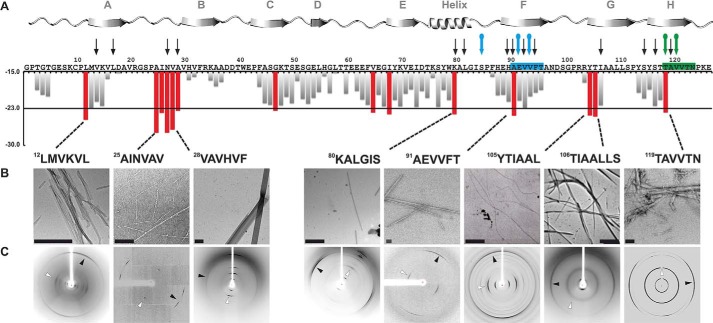
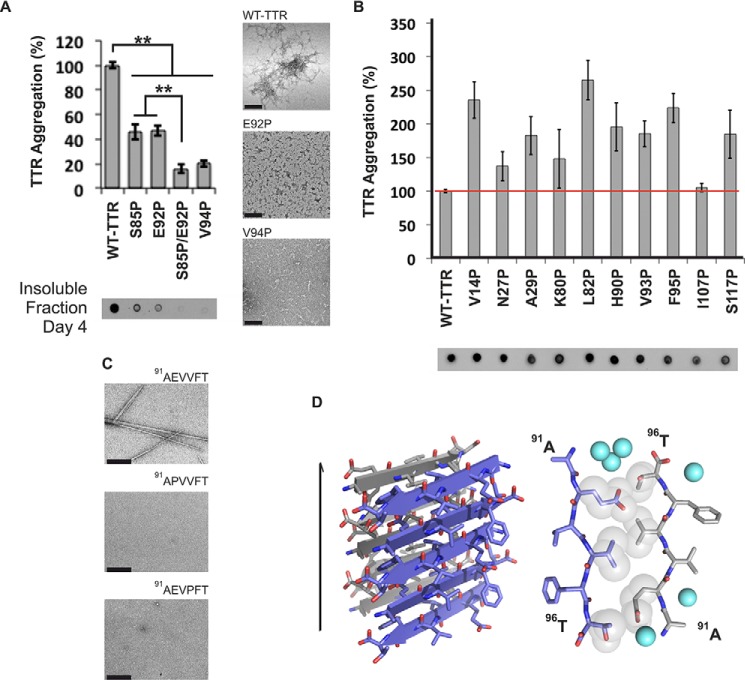
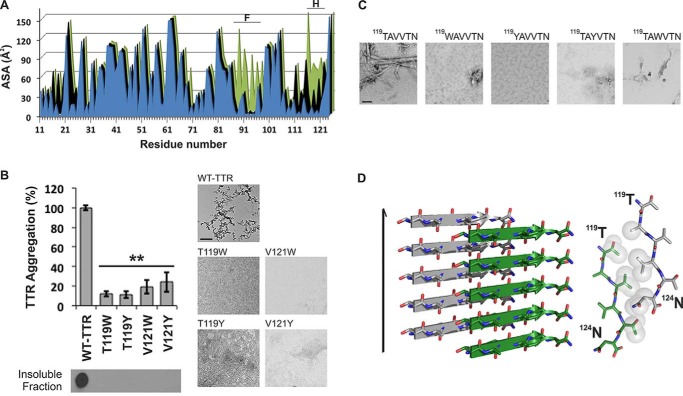
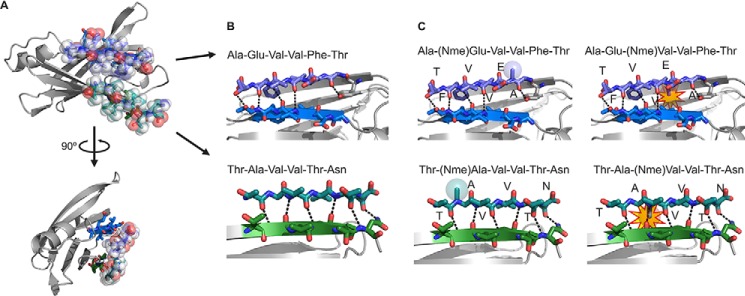
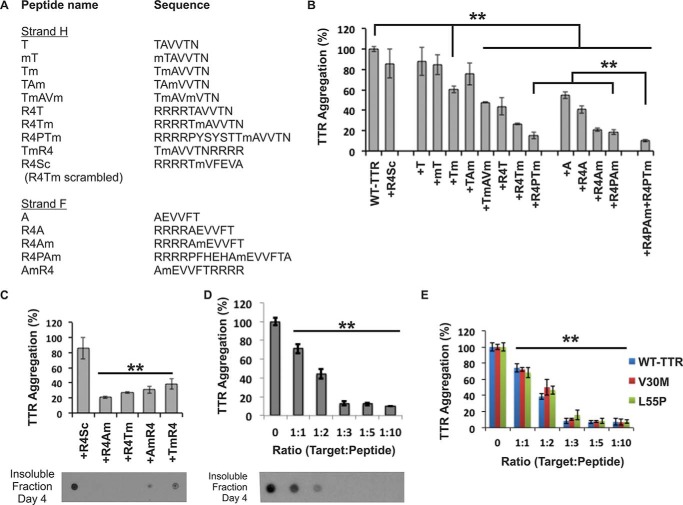
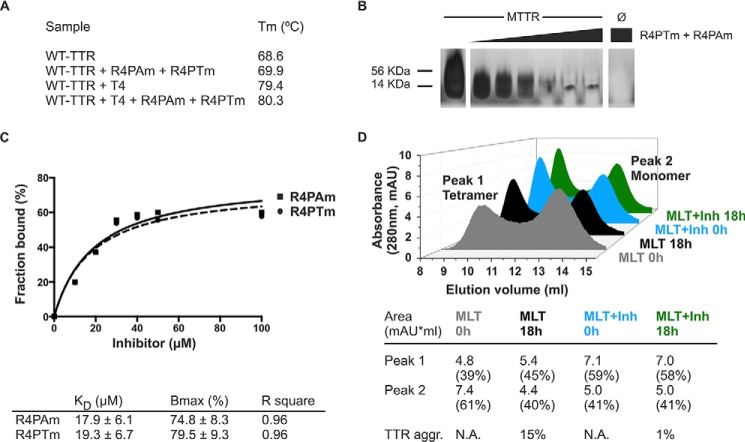
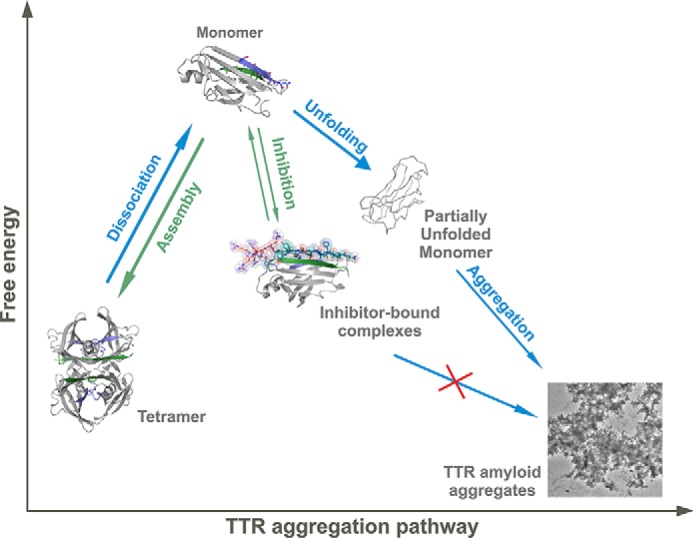
The tetrameric thyroxine transport protein transthyretin (TTR) forms amyloid fibrils upon dissociation and monomer unfolding. The aggregation of transthyretin has been reported as the cause of the life-threatening transthyretin amyloidosis. The standard treatment of familial cases of TTR amyloidosis has been liver transplantation. Although aggregation-preventing strategies involving ligands are known, understanding the mechanism of TTR aggregation can lead to additional inhibition approaches. Several models of TTR amyloid fibrils have been proposed, but the segments that drive aggregation of the protein have remained unknown. Here we identify β-strands F and H as necessary for TTR aggregation. Based on the crystal structures of these segments, we designed two non-natural peptide inhibitors that block aggregation. This work provides the first characterization of peptide inhibitors for TTR aggregation, establishing a novel therapeutic strategy.
Keywords: TTR; amyloid; inhibition mechanism; mutational analysis; peptide interaction; protein aggregation; transthyretin amyloidosis; x-ray crystallography.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
A pair of peptides inhibits seeding of the hormone transporter transthyretin into amyloid fibrils.J Biol Chem. 2019 Apr 12;294(15):6130-6141. doi: 10.1074/jbc.RA118.005257. Epub 2019 Feb 7. J Biol Chem. 2019. PMID: 30733338 Free PMC article.
-
Tolcapone, a potent aggregation inhibitor for the treatment of familial leptomeningeal amyloidosis.FEBS J. 2021 Jan;288(1):310-324. doi: 10.1111/febs.15339. Epub 2020 May 11. FEBS J. 2021. PMID: 32324953
-
Specific disruption of transthyretin(105-115) fibrilization using "stabilizing" inhibitors of transthyretin amyloidogenesis.Biochemistry. 2012 Apr 24;51(16):3523-30. doi: 10.1021/bi3002727. Epub 2012 Apr 12. Biochemistry. 2012. PMID: 22482799
-
Review on the Structures and Activities of Transthyretin Amyloidogenesis Inhibitors.Drug Des Devel Ther. 2020 Mar 10;14:1057-1081. doi: 10.2147/DDDT.S237252. eCollection 2020. Drug Des Devel Ther. 2020. PMID: 32210536 Free PMC article. Review.
-
Uncovering the Neuroprotective Mechanisms of Curcumin on Transthyretin Amyloidosis.Int J Mol Sci. 2019 Mar 14;20(6):1287. doi: 10.3390/ijms20061287. Int J Mol Sci. 2019. PMID: 30875761 Free PMC article. Review.
Cited by
-
Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study.Eur J Heart Fail. 2021 Feb;23(2):277-285. doi: 10.1002/ejhf.2027. Epub 2020 Nov 12. Eur J Heart Fail. 2021. PMID: 33070419 Free PMC article. Clinical Trial.
-
A crystal-structural study of Pauling-Corey rippled sheets.Chem Sci. 2021 Dec 8;13(3):671-680. doi: 10.1039/d1sc05731f. eCollection 2022 Jan 19. Chem Sci. 2021. PMID: 35173931 Free PMC article.
-
Transthyretin Aggregation Pathway toward the Formation of Distinct Cytotoxic Oligomers.Sci Rep. 2019 Jan 10;9(1):33. doi: 10.1038/s41598-018-37230-1. Sci Rep. 2019. PMID: 30631096 Free PMC article.
-
The Regulatory Mechanism of Transthyretin Irreversible Aggregation through Liquid-to-Solid Phase Transition.Int J Mol Sci. 2023 Feb 13;24(4):3729. doi: 10.3390/ijms24043729. Int J Mol Sci. 2023. PMID: 36835140 Free PMC article.
-
Defensin-based therapeutic peptide design in attenuating V30M TTR-induced Familial Amyloid Polyneuropathy.3 Biotech. 2023 Jul;13(7):227. doi: 10.1007/s13205-023-03646-4. Epub 2023 Jun 8. 3 Biotech. 2023. PMID: 37304406 Free PMC article.
References
-
- Chung C. M., Connors L. H., Benson M. D., and Walsh M. T. (2001) Biophysical analysis of normal transthyretin: implications for fibril formation in senile systemic amyloidosis. Amyloid 8, 75–83 - PubMed
-
- Colon W., and Kelly J. W. (1992) Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 31, 8654–8660 - PubMed
-
- Foss T. R., Wiseman R. L., and Kelly J. W. (2005) The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry 44, 15525–15533 - PubMed
-
- Lai Z., Colón W., and Kelly J. W. (1996) The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 35, 6470–6482 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
