

Peroxisome proliferator-activated receptor-α-mediated transcription of miR-301a and miR-454 and their host gene SKA2 regulates endothelin-1 and PAI-1 expression in sickle cell disease
- PMID: 26460070
- PMCID: PMC4672349
- DOI: 10.1042/BSR20150190
Peroxisome proliferator-activated receptor-α-mediated transcription of miR-301a and miR-454 and their host gene SKA2 regulates endothelin-1 and PAI-1 expression in sickle cell disease
Abstract
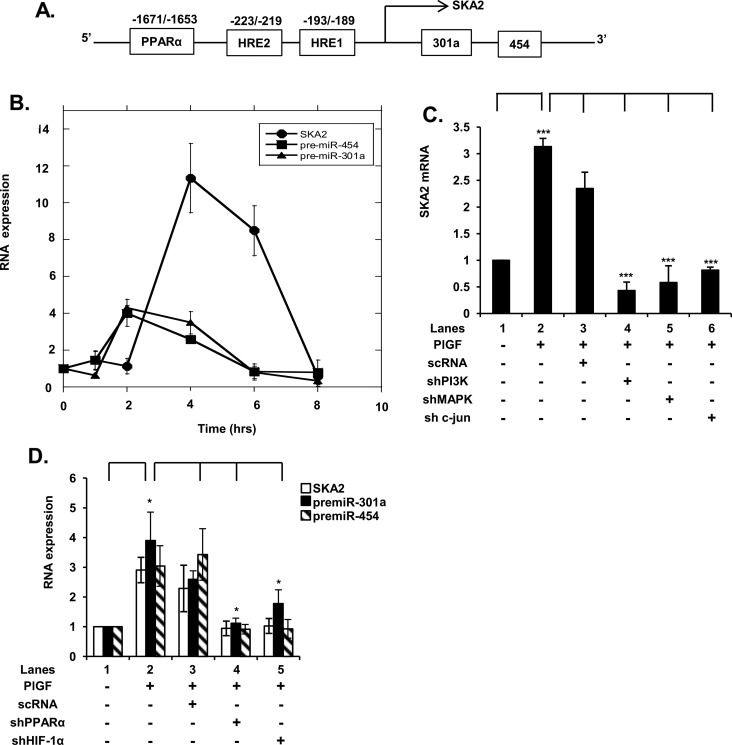
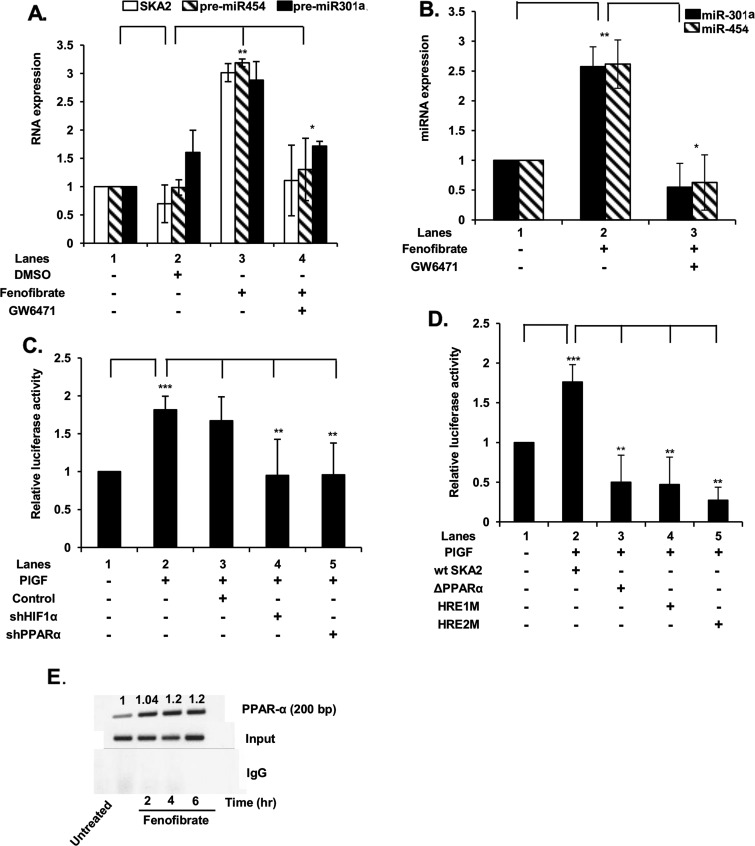
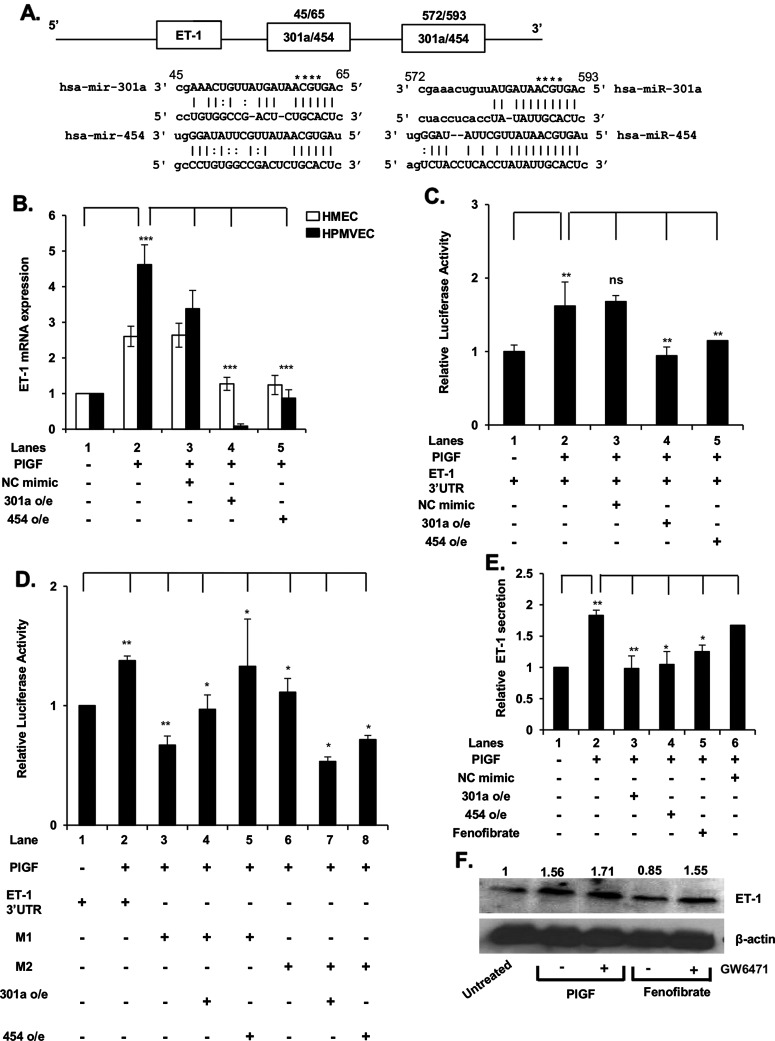
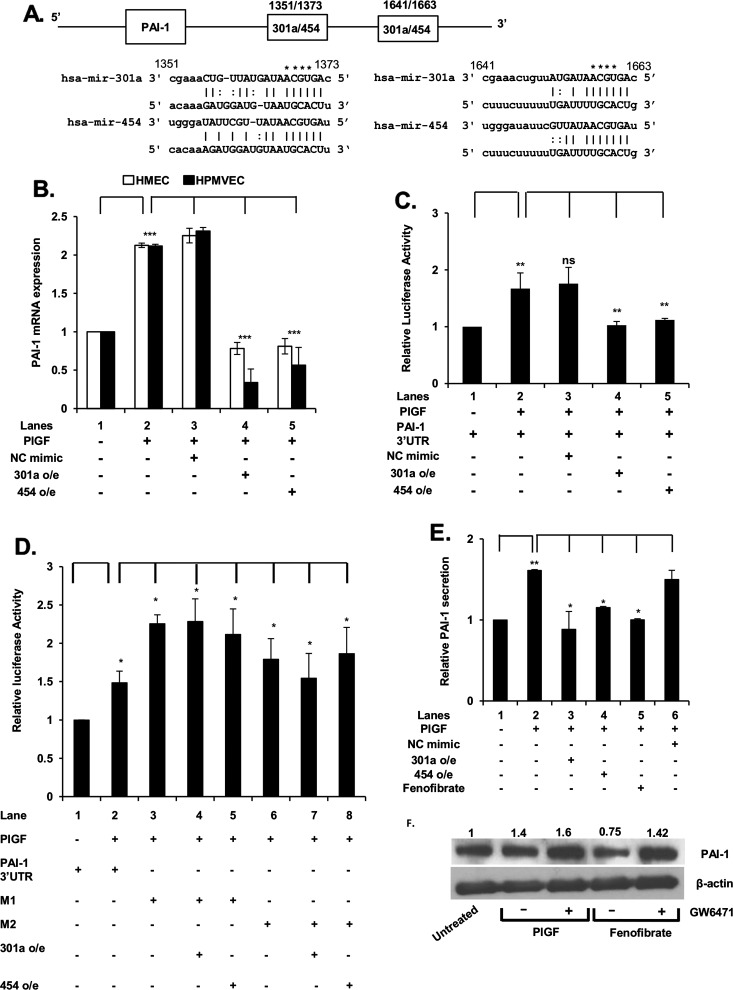
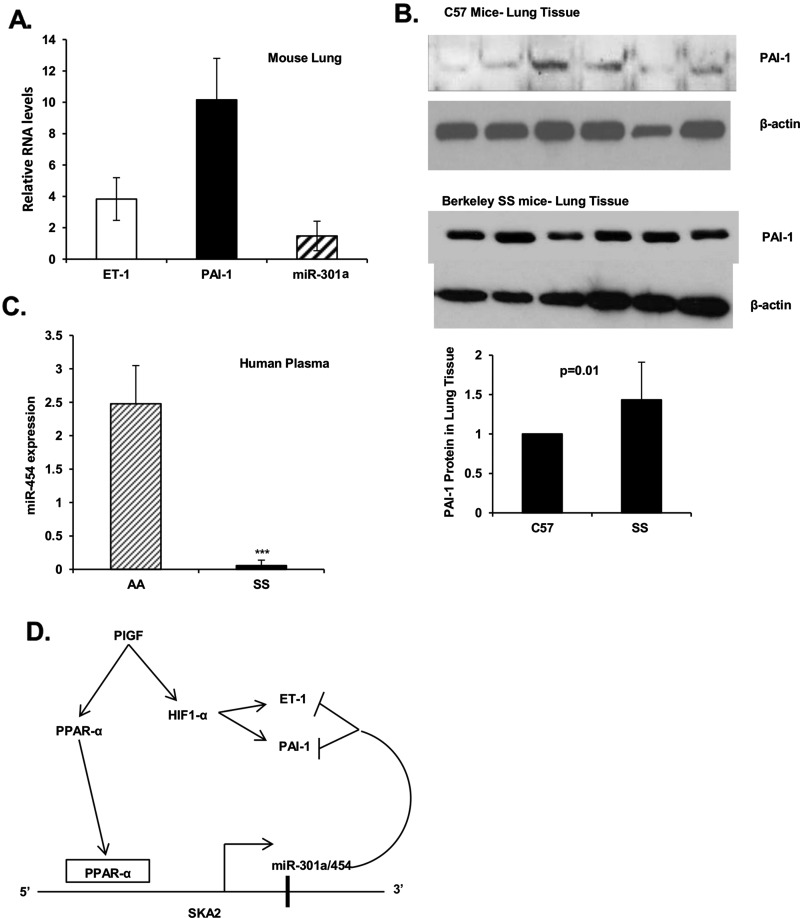
Endothelin-1 (ET-1) and plasminogen activator inhibitor-1 (PAI-1) play important roles in pulmonary hypertension (PH) in sickle cell disease (SCD). Our previous studies show higher levels of placenta growth factor (PlGF) in SCD correlate with increased plasma levels of ET-1, PAI-1, and other physiological markers of PH. PlGF-mediated ET-1 and PAI-1 expression occurs via activation of hypoxia-inducible factor-1α (HIF-1α). However, relatively little is understood regarding post-transcriptional regulation of PlGF-mediated expression of ET-1 and PAI-1. Herein, we show PlGF treatment of endothelial cells reduced levels of miR-301a and miR-454 from basal levels. In addition, both miRNAs targeted the 3'-UTRs of ET-1 and PAI-1 mRNAs. These results were corroborated in the mouse model of SCD [Berkeley sickle mice (BK-SS)] and in SCD subjects. Plasma levels of miR-454 in SCD subjects were significantly lower compared with unaffected controls, which correlated with higher plasma levels of both ET-1 and PAI-1. Moreover, lung tissues from BK-SS mice showed significantly reduced levels of pre-miR-301a and concomitantly higher levels of ET-1 and PAI-1. Furthermore, we show that miR-301a/miR-454 located in the spindle and kinetochore-associated protein-2 (SKA2) transcription unit was co-transcriptionally regulated by both HIF-1α and peroxisome proliferator-activated receptor-α (PPAR-α) as demonstrated by SKA2 promoter mutational analysis and ChIP. Finally we show that fenofibrate, a PPAR-α agonist, increased the expression of miR-301a/miR-454 and SKA2 in human microvascular endothelial cell line (HMEC) cells; the former were responsible for reduced expression of ET-1 and PAI-1. Our studies provide a potential therapeutic approach whereby fenofibrate-induced miR-301a/miR-454 expression can ameliorate PH and lung fibrosis by reduction in ET-1 and PAI-1 levels in SCD.
Keywords: endothelin-1 (ET-1); micro ribonucleic acid (miRNA); peroxisome proliferator-activated receptor-α (PPAR-α); plasminogen activator inhibitor-1 (PAI-1); sickle cell disease (SCD); spindle and kinetochore-associated protein-2 (SKA2).
© 2015 Authors.
Figures
Similar articles
-
Peroxisome proliferator-activated receptor-α-mediated transcription of miR-199a2 attenuates endothelin-1 expression via hypoxia-inducible factor-1α.J Biol Chem. 2014 Dec 26;289(52):36031-47. doi: 10.1074/jbc.M114.600775. Epub 2014 Nov 11. J Biol Chem. 2014. PMID: 25389292 Free PMC article.
-
Erythropoietin-mediated expression of placenta growth factor is regulated via activation of hypoxia-inducible factor-1α and post-transcriptionally by miR-214 in sickle cell disease.Biochem J. 2015 Jun 15;468(3):409-23. doi: 10.1042/BJ20141138. Epub 2015 Apr 16. Biochem J. 2015. PMID: 25876995 Free PMC article.
-
Involvement of miR-30c and miR-301a in immediate induction of plasminogen activator inhibitor-1 by placental growth factor in human pulmonary endothelial cells.Biochem J. 2011 Mar 15;434(3):473-82. doi: 10.1042/BJ20101585. Biochem J. 2011. PMID: 21175428 Free PMC article.
-
Placenta growth factor mediated gene regulation in sickle cell disease.Blood Rev. 2018 Jan;32(1):61-70. doi: 10.1016/j.blre.2017.08.008. Epub 2017 Aug 16. Blood Rev. 2018. PMID: 28823762 Free PMC article. Review.
-
Regulation of the MIR155 host gene in physiological and pathological processes.Gene. 2013 Dec 10;532(1):1-12. doi: 10.1016/j.gene.2012.12.009. Epub 2012 Dec 14. Gene. 2013. PMID: 23246696 Review.
Cited by
-
A Pilot Study on MicroRNA Profile in Tear Fluid to Predict Response to Anti-VEGF Treatments for Diabetic Macular Edema.J Clin Med. 2020 Sep 10;9(9):2920. doi: 10.3390/jcm9092920. J Clin Med. 2020. PMID: 32927780 Free PMC article.
-
Polymorphism of the endothelin-1 gene (rs5370) is a potential contributor to sickle cell disease pathophysiology.Genes Dis. 2016 Sep 30;3(4):294-298. doi: 10.1016/j.gendis.2016.09.002. eCollection 2016 Dec. Genes Dis. 2016. PMID: 30258900 Free PMC article.
-
The Worst Things in Life are Free: The Role of Free Heme in Sickle Cell Disease.Front Immunol. 2021 Jan 27;11:561917. doi: 10.3389/fimmu.2020.561917. eCollection 2020. Front Immunol. 2021. PMID: 33584641 Free PMC article. Review.
-
Inhibition of miR301 enhances Akt-mediated cell proliferation by accumulation of PTEN in nucleus and its effects on cell-cycle regulatory proteins.Oncotarget. 2016 Apr 12;7(15):20953-65. doi: 10.18632/oncotarget.7996. Oncotarget. 2016. PMID: 26967567 Free PMC article.
-
The inflammatory profiles of pulmonary alveolar macrophages and alveolar type 2 cells in SCD.Exp Biol Med (Maywood). 2023 Jun;248(12):1013-1023. doi: 10.1177/15353702231157940. Epub 2023 Apr 3. Exp Biol Med (Maywood). 2023. PMID: 37012678 Free PMC article.
References
-
- Hsu L.L., Champion H.C., Campbell-Lee S.A., Bivalacqua T.J., Manci E.A., Diwan B.A., Schimel D.M., Cochard A.E., Wang X., Schechter A.N., et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood. 2007;109:3088–3098. doi: 10.1182/blood-2006-06-029173. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous