

Cell(s) of Origin of Langerhans Cell Histiocytosis
- PMID: 26461145
- PMCID: PMC4699587
- DOI: 10.1016/j.hoc.2015.06.003
Cell(s) of Origin of Langerhans Cell Histiocytosis
Abstract
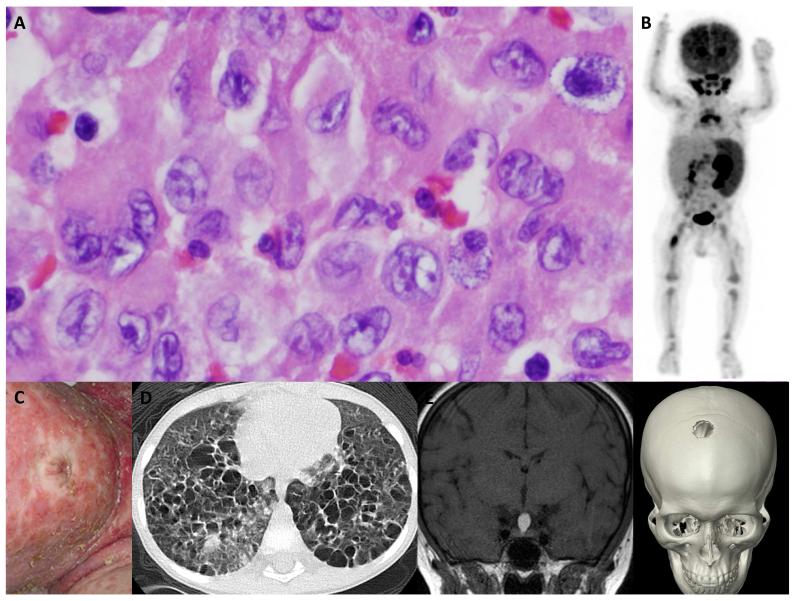
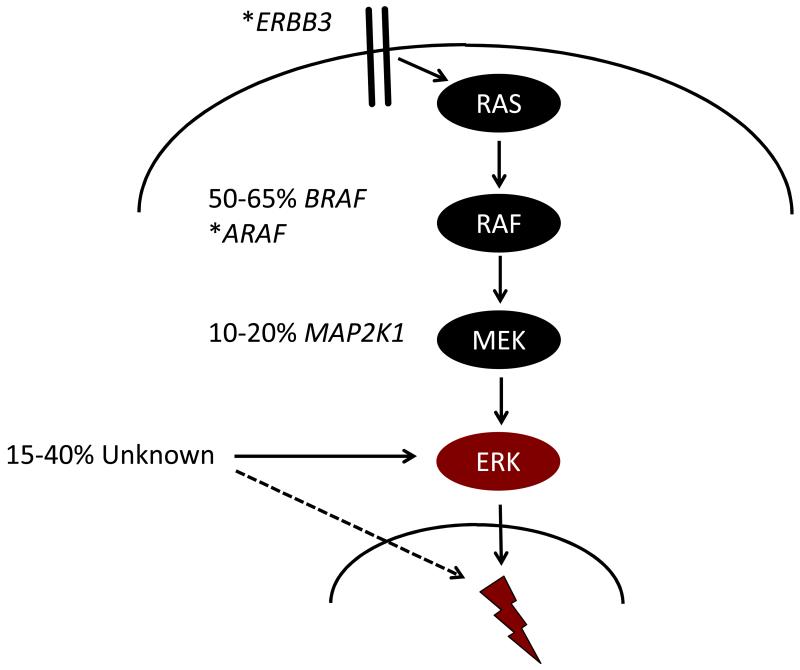
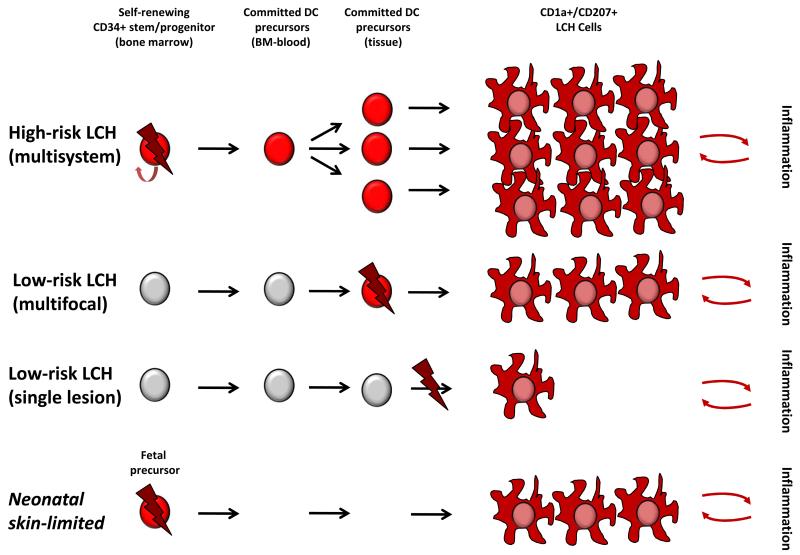
Langerhans cell histiocytosis (LCH) is heterogeneous disease characterized by common histology of inflammatory lesions containing Langerin(+) (CD207) histiocytes. Emerging data support a model in which MAPK activation in self-renewing hematopoietic progenitors may drive disseminated high-risk disease, whereas MAPK activation in more differentiated committed myeloid populations may induce low-risk LCH. The heterogeneous clinical manifestations with shared histology may represent the final common pathway of an acquired defect of differentiation, initiated at more than one point. Implications of this model include re-definition of LCH as a myeloid neoplasia and re-focusing therapeutic strategies on the cells and lineages of origin.
Keywords: BRAF; Dendritic cell; Langerhans cell histiocytosis; MAPK signaling; Myeloid differentiation.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
References
-
- Nezelof C, Basset F, Rousseau MF. Histiocytosis × histogenetic arguments for a Langerhans cell origin. Biomedicine. 1973;18:365–371. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
