

Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors
- PMID: 26463424
- PMCID: PMC4705604
- DOI: 10.1182/blood-2015-06-654194
Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors
Abstract
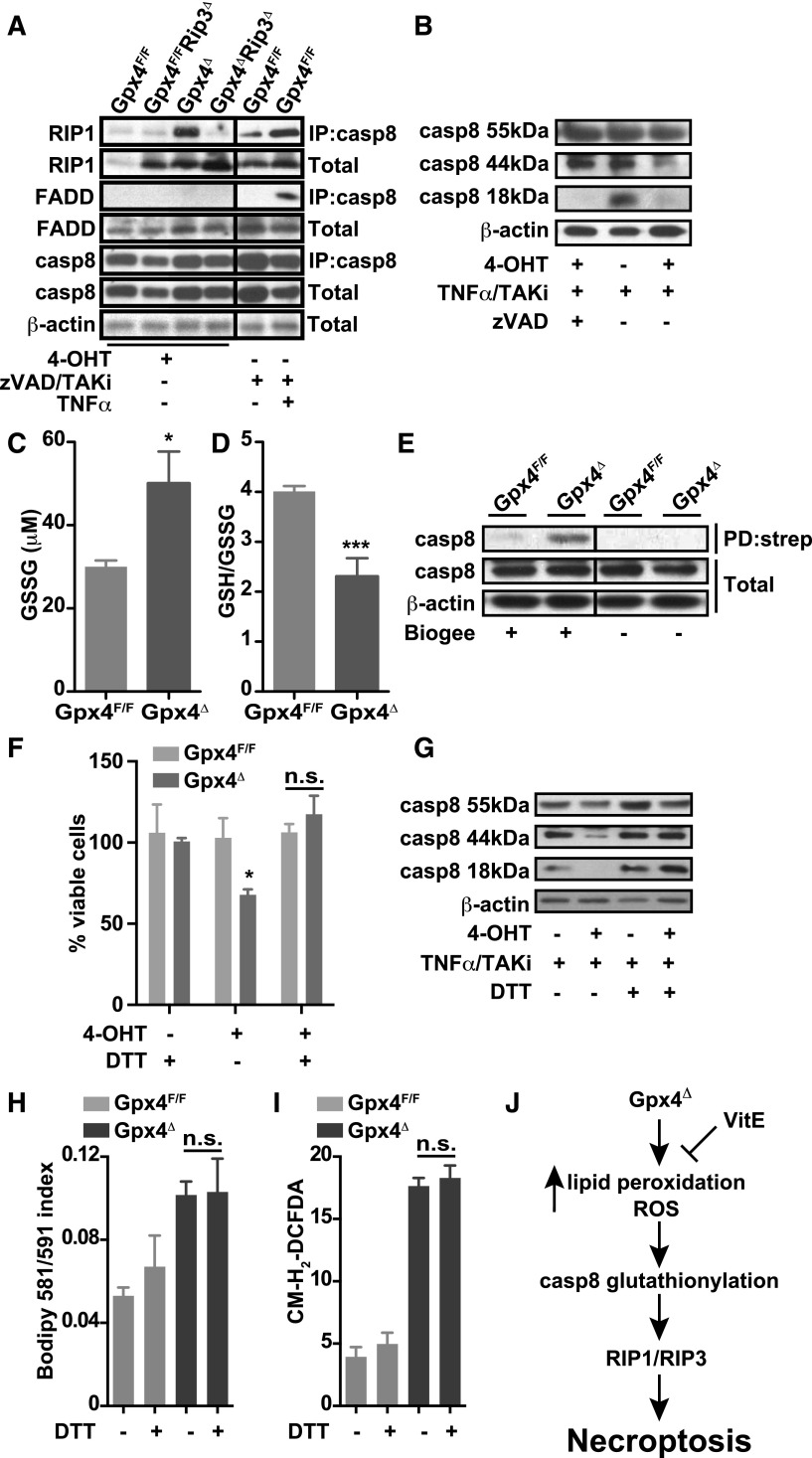
Maintaining cellular redox balance is vital for cell survival and tissue homoeostasis because imbalanced production of reactive oxygen species (ROS) may lead to oxidative stress and cell death. The antioxidant enzyme glutathione peroxidase 4 (Gpx4) is a key regulator of oxidative stress-induced cell death. We show that mice with deletion of Gpx4 in hematopoietic cells develop anemia and that Gpx4 is essential for preventing receptor-interacting protein 3 (RIP3)-dependent necroptosis in erythroid precursor cells. Absence of Gpx4 leads to functional inactivation of caspase 8 by glutathionylation, resulting in necroptosis, which occurs independently of tumor necrosis factor α activation. Although genetic ablation of Rip3 normalizes reticulocyte maturation and prevents anemia, ROS accumulation and lipid peroxidation in Gpx4-deficient cells remain high. Our results demonstrate that ROS and lipid hydroperoxides function as not-yet-recognized unconventional upstream signaling activators of RIP3-dependent necroptosis.
© 2016 by The American Society of Hematology.
Figures






Comment in
-
Unconventional cell death in erythroid cells.Blood. 2016 Jan 7;127(1):12-4. doi: 10.1182/blood-2015-11-677229. Blood. 2016. PMID: 26744438 No abstract available.
References
-
- D’Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813–824. - PubMed
-
- Vanden Berghe T, Vanlangenakker N, Parthoens E, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17(6):922–930. - PubMed
-
- Fiers W, Beyaert R, Declercq W, Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18(54):7719–7730. - PubMed
-
- Beutler E, Luzzatto L. Hemolytic anemia. Semin Hematol. 1999;36(4 Suppl 7):38–47. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
