

Building an RNA Sequencing Transcriptome of the Central Nervous System
- PMID: 26463470
- PMCID: PMC4833695
- DOI: 10.1177/1073858415610541
Building an RNA Sequencing Transcriptome of the Central Nervous System
Abstract
The composition and function of the central nervous system (CNS) is extremely complex. In addition to hundreds of subtypes of neurons, other cell types, including glia (astrocytes, oligodendrocytes, and microglia) and vascular cells (endothelial cells and pericytes) also play important roles in CNS function. Such heterogeneity makes the study of gene transcription in CNS challenging. Transcriptomic studies, namely the analyses of the expression levels and structures of all genes, are essential for interpreting the functional elements and understanding the molecular constituents of the CNS. Microarray has been a predominant method for large-scale gene expression profiling in the past. However, RNA-sequencing (RNA-Seq) technology developed in recent years has many advantages over microarrays, and has enabled building more quantitative, accurate, and comprehensive transcriptomes of the CNS and other systems. The discovery of novel genes, diverse alternative splicing events, and noncoding RNAs has remarkably expanded the complexity of gene expression profiles and will help us to understand intricate neural circuits. Here, we discuss the procedures and advantages of RNA-Seq technology in mammalian CNS transcriptome construction, and review the approaches of sample collection as well as recent progress in building RNA-Seq-based transcriptomes from tissue samples and specific cell types.
Keywords: RNA-sequencing; alternative splicing; central nervous system; gene expression; noncoding RNA; transcriptome complexity.
© The Author(s) 2015.
Conflict of interest statement
Declaration of Conflicting Interests The author(s) declared no potential conflict of interest with respect to the research, authorship, and/or publication of this article.
Figures
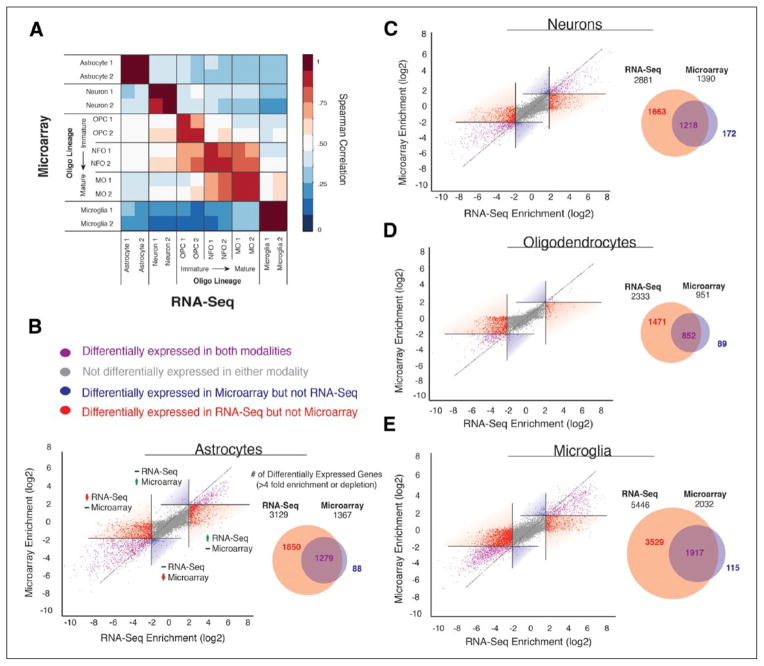
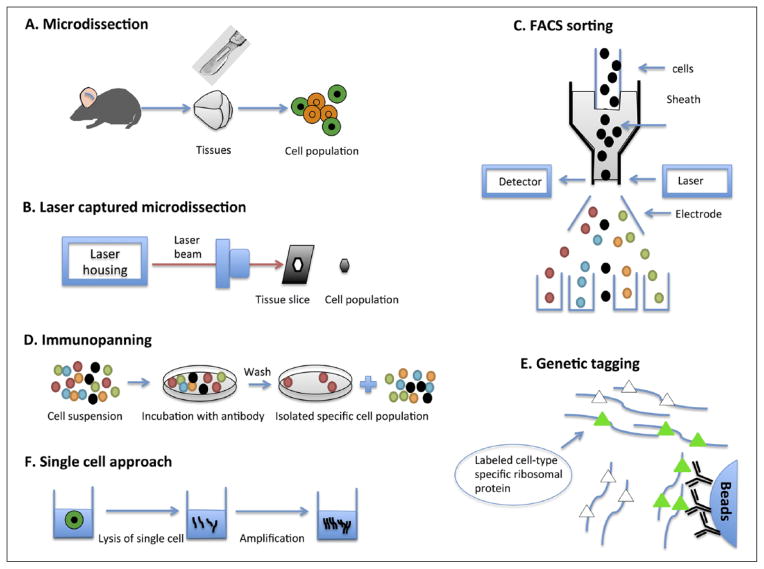
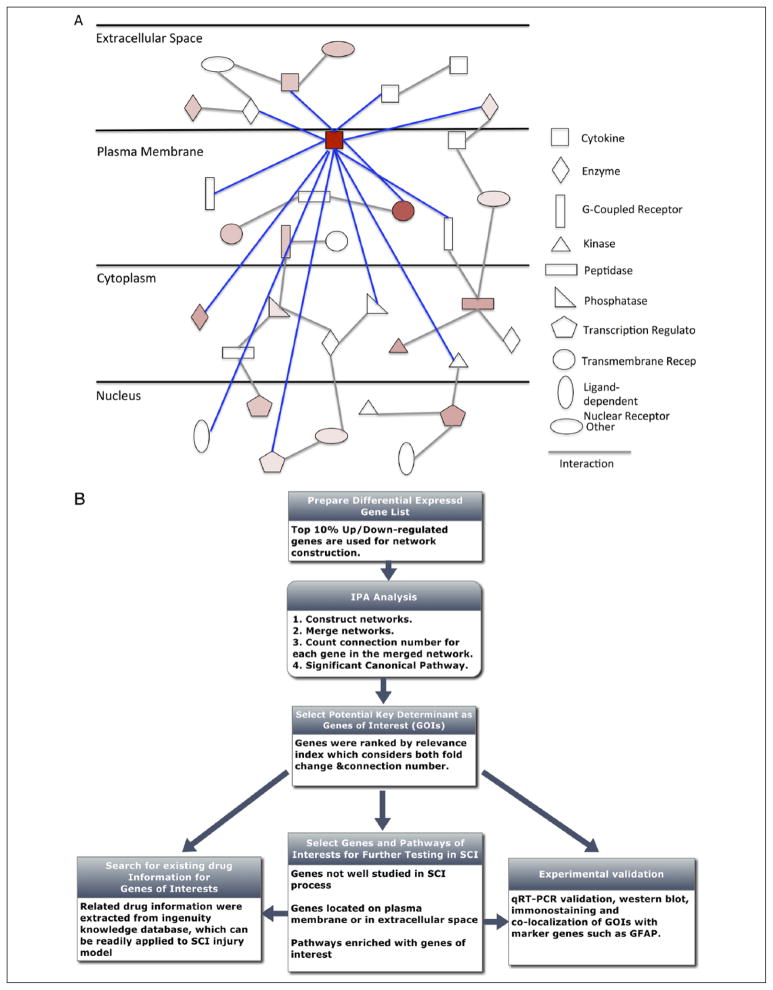
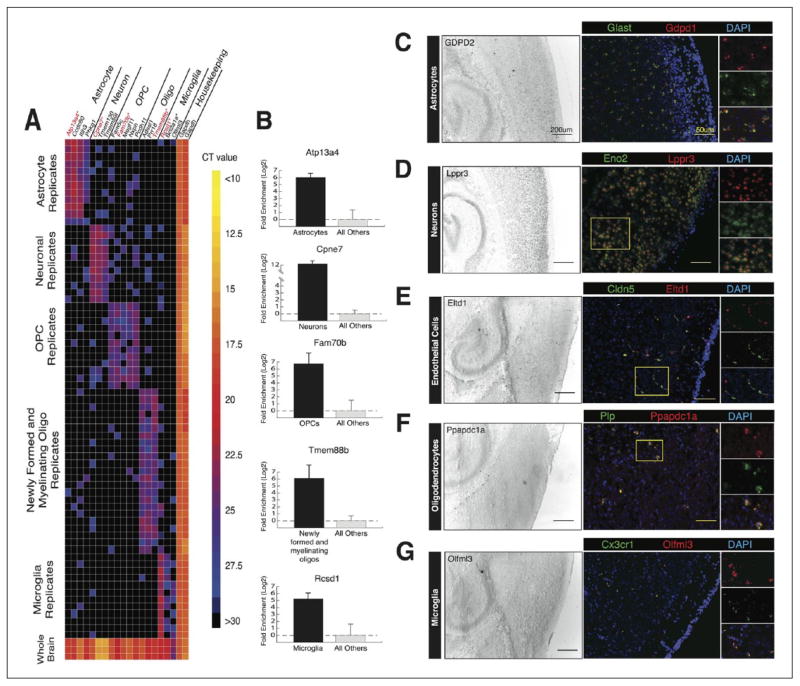
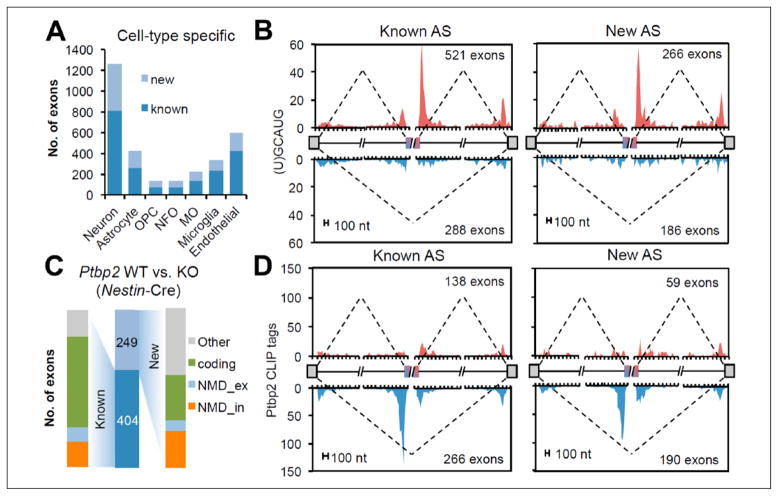
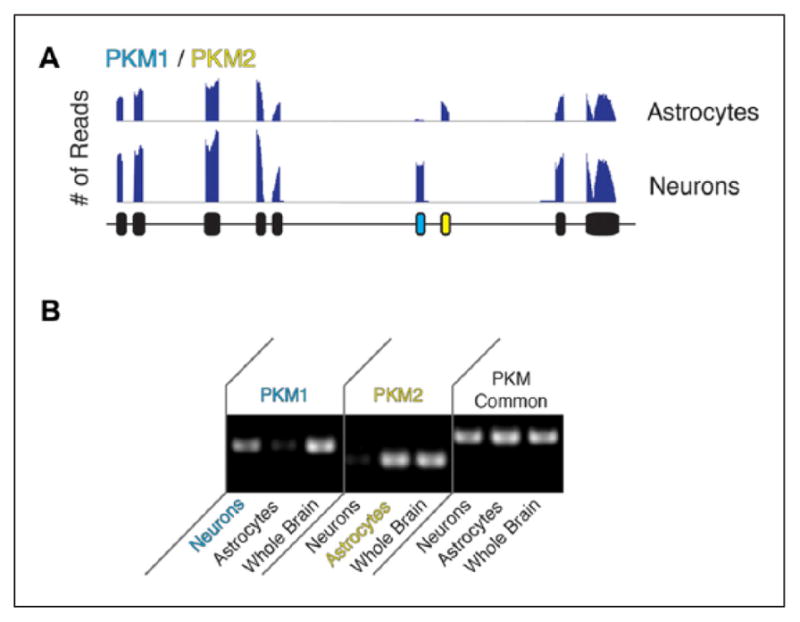
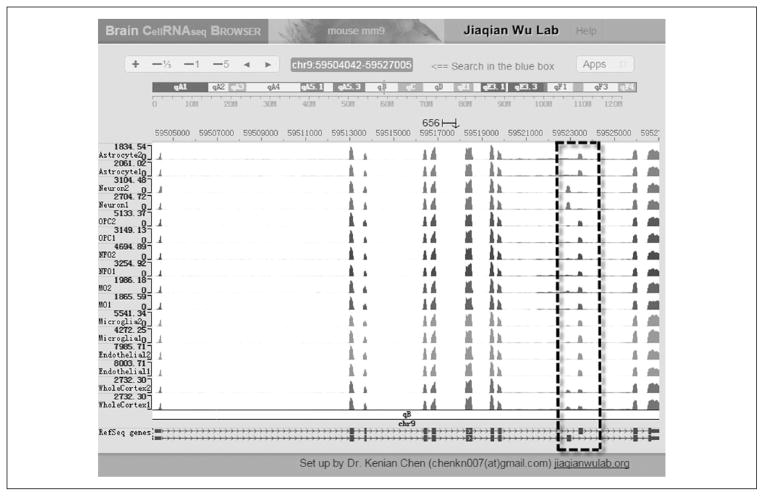
References
-
- Bareyre FM, Schwab ME. Inflammation, degeneration and regeneration in the injured spinal cord: insights from DNA microarrays. Trends Neurosci. 2003;26(10):555–63. - PubMed
-
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, et al. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70(1):31–46. - PubMed
-
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, et al. Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nat Genet. 2001;29(4):365–71. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
