

PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction
- PMID: 26464484
- PMCID: PMC4664165
- DOI: 10.1093/hmg/ddv423
PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction
Abstract
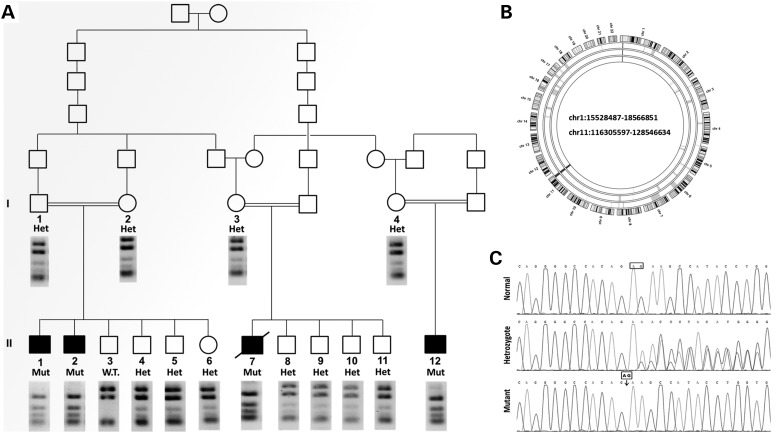
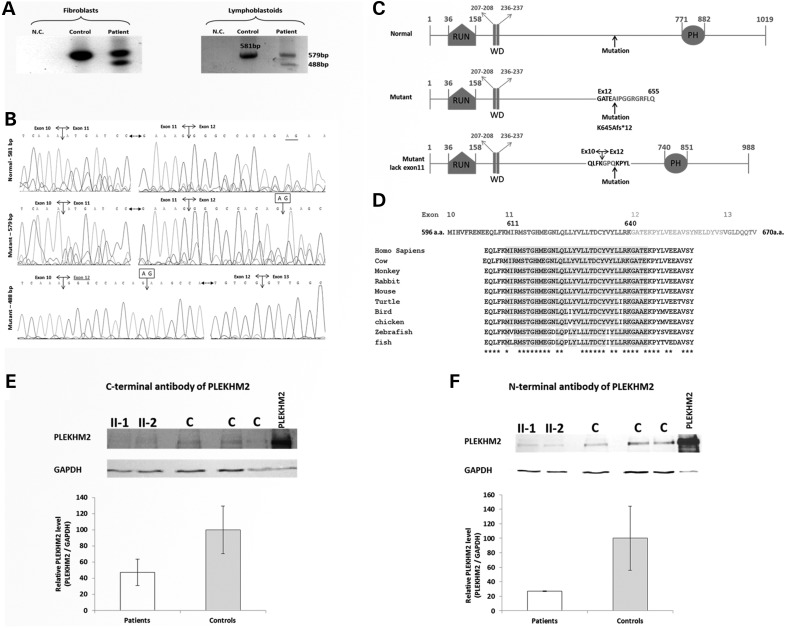
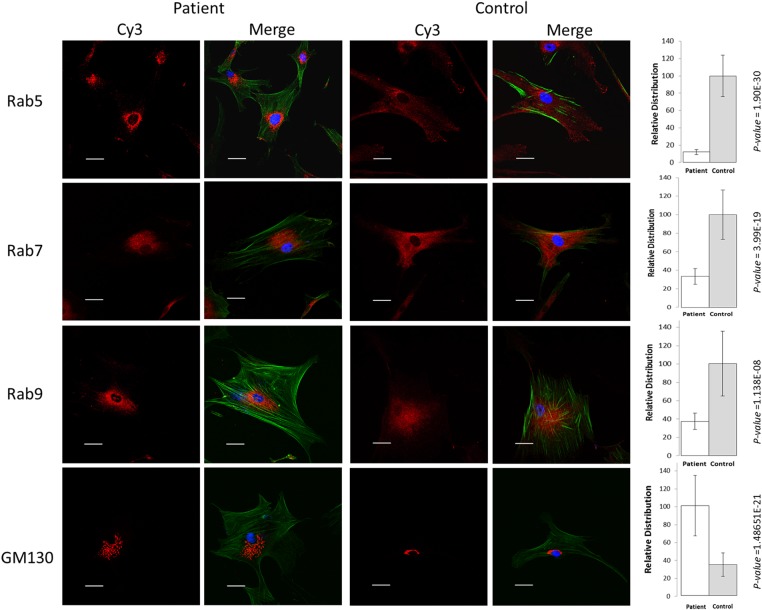
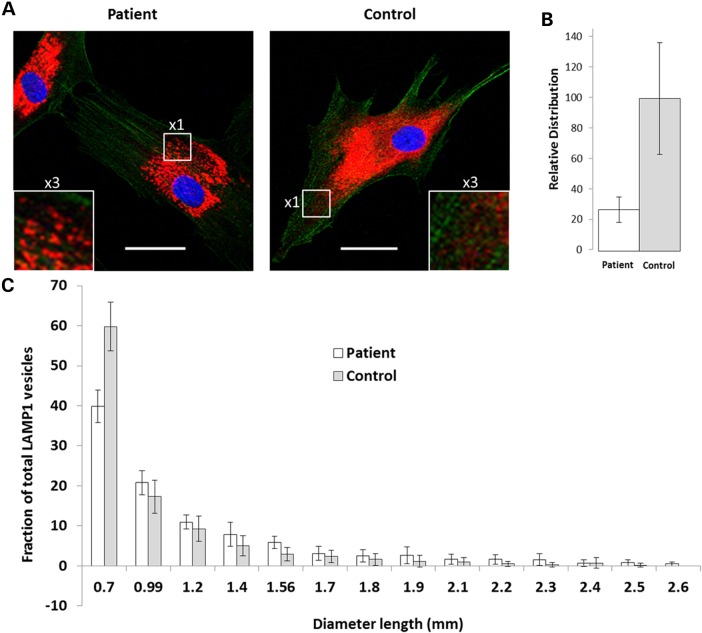
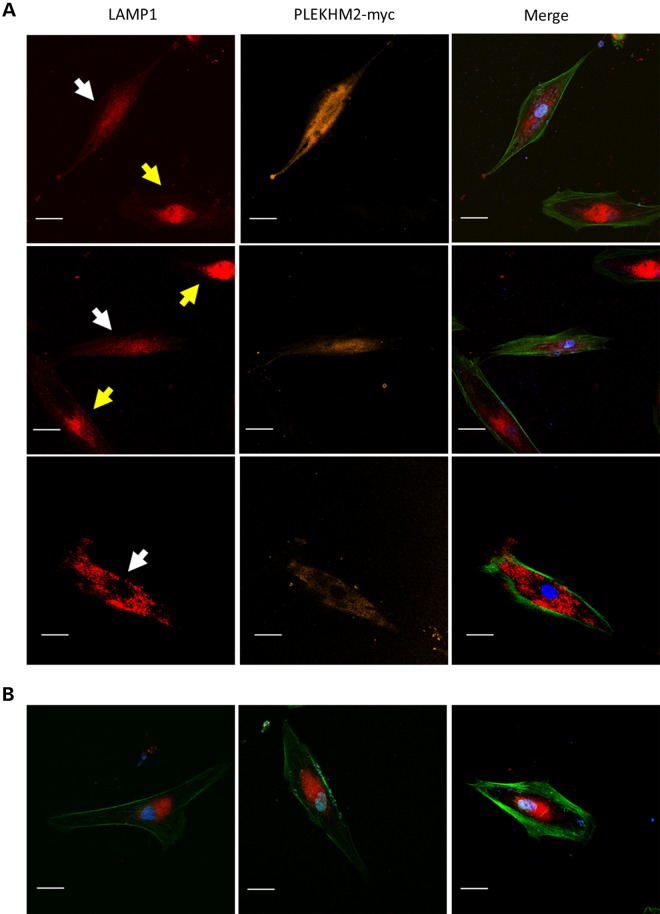
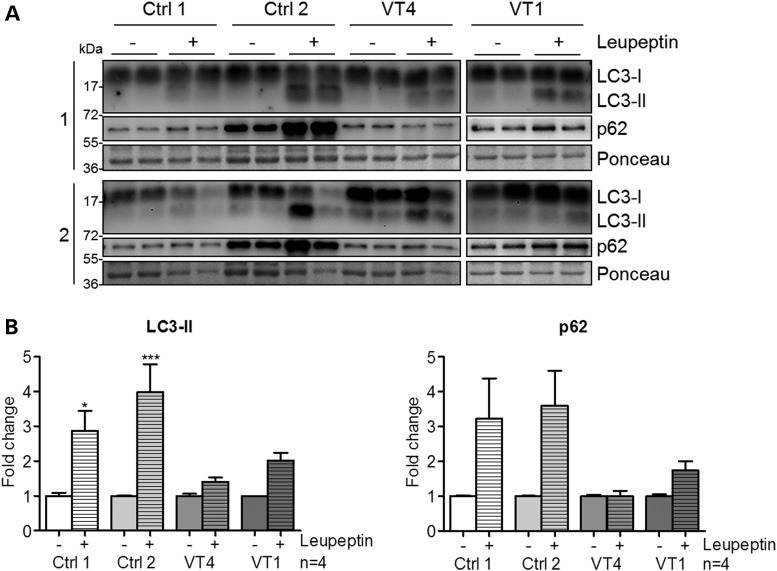
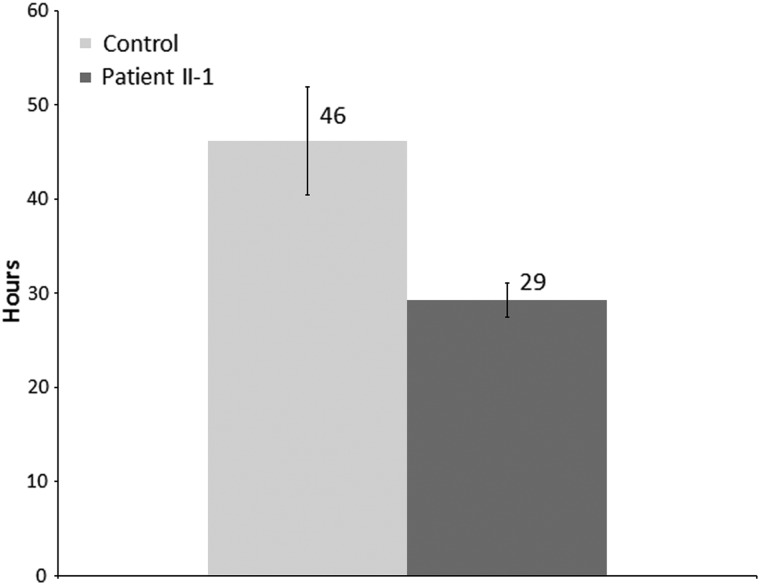
Gene mutations, mostly segregating with a dominant mode of inheritance, are important causes of dilated cardiomyopathy (DCM), a disease characterized by enlarged ventricular dimensions, impaired cardiac function, heart failure and high risk of death. Another myocardial abnormality often linked to gene mutations is left ventricular noncompaction (LVNC) characterized by a typical diffuse spongy appearance of the left ventricle. Here, we describe a large Bedouin family presenting with a severe recessive DCM and LVNC. Homozygosity mapping and exome sequencing identified a single gene variant that segregated as expected and was neither reported in databases nor in Bedouin population controls. The PLEKHM2 cDNA2156_2157delAG variant causes the frameshift p.Lys645AlafsTer12 and/or the skipping of exon 11 that results in deletion of 30 highly conserved amino acids. PLEKHM2 is known to interact with several Rabs and with kinesin-1, affecting endosomal trafficking. Accordingly, patients' primary fibroblasts exhibited abnormal subcellular distribution of endosomes marked by Rab5, Rab7 and Rab9, as well as the Golgi apparatus. In addition, lysosomes appeared to be concentrated in the perinuclear region, and autophagy flux was impaired. Transfection of wild-type PLEKHM2 cDNA into patient's fibroblasts corrected the subcellular distribution of the lysosomes, supporting the causal effect of PLEKHM2 mutation. PLEKHM2 joins LAMP-2 and BAG3 as a disease gene altering autophagy resulting in an isolated cardiac phenotype. The association of PLEKHM2 mutation with DCM and LVNC supports the importance of autophagy for normal cardiac function.
© The Author 2015. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Yancy C.W., Jessup M., Bozkurt B., Butler J., Casey D.E. Jr, Drazner M.H., Fonarow G.C., Geraci S.A., Horwich T., Januzzi J.L. et al. (2013) 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation, 128, e240–e327. - PubMed
-
- McMurray J.J., Adamopoulos S., Anker S.D., Auricchio A., Bohm M., Dickstein K., Falk V., Filippatos G., Fonseca C., Gomez-Sanchez M.A. et al. (2012) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the task force for the diagnosis and treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail., 14, 803–869. - PubMed
-
- Piran S., Liu P., Morales A., Hershberger R.E. (2012) Where genome meets phenome: rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J. Am. Coll. Cardiol., 60, 283–289. - PubMed
-
- Gregori D., Rocco C., Miocic S., Mestroni L. (2001) Estimating the frequency of familial dilated cardiomyopathy in the presence of misclassification errors. J. Appl. Stat., 28, 53–62.
-
- Petretta M., Pirozzi F., Sasso L., Paglia A., Bonaduce D. (2011) Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am. J. Cardiol., 108, 1171–1176. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
