

Individual Cytokines Modulate the Neurological Symptoms of ATM Deficiency in a Region Specific Manner
- PMID: 26465009
- PMCID: PMC4596028
- DOI: 10.1523/ENEURO.0032-15.2015
Individual Cytokines Modulate the Neurological Symptoms of ATM Deficiency in a Region Specific Manner
Abstract
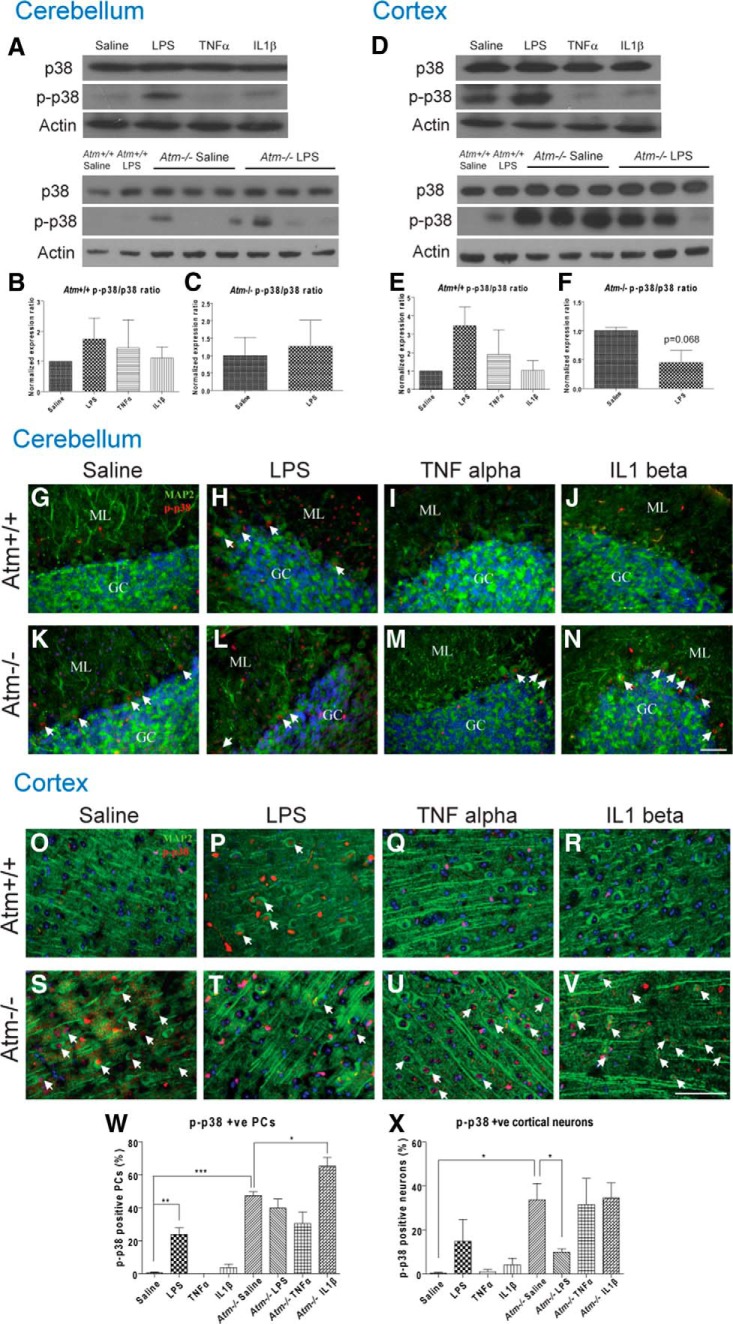
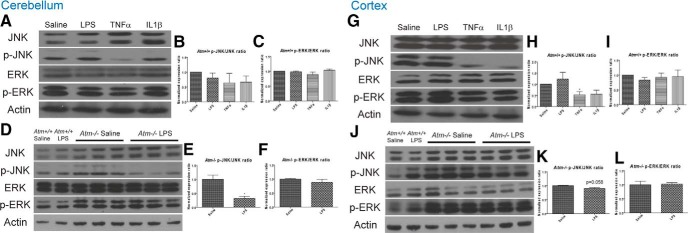
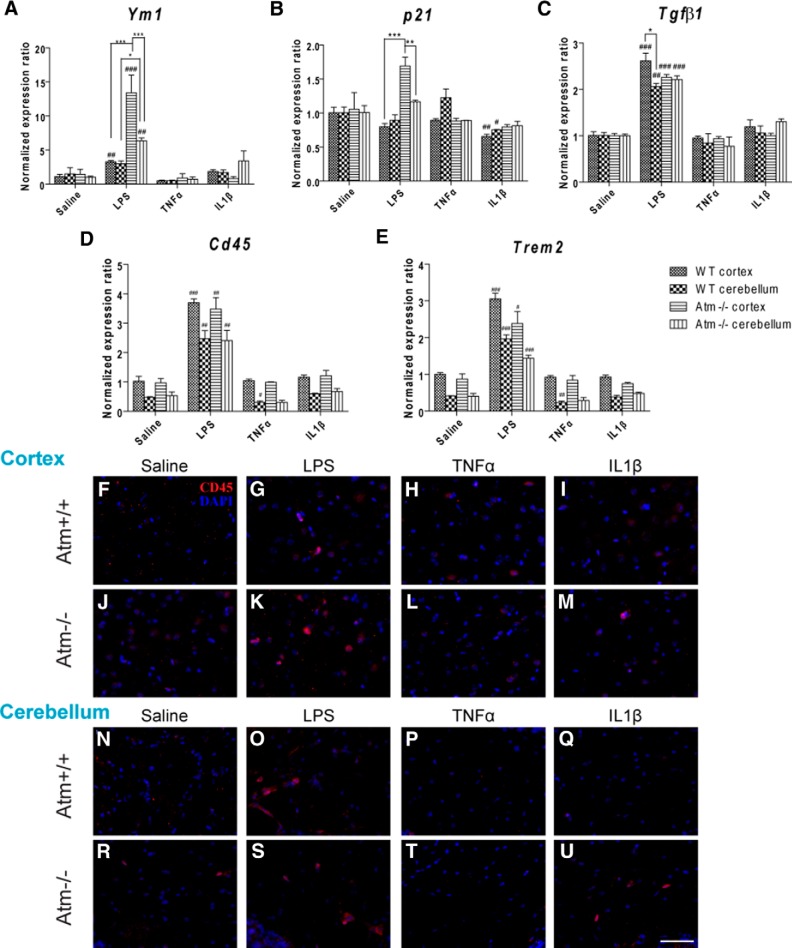
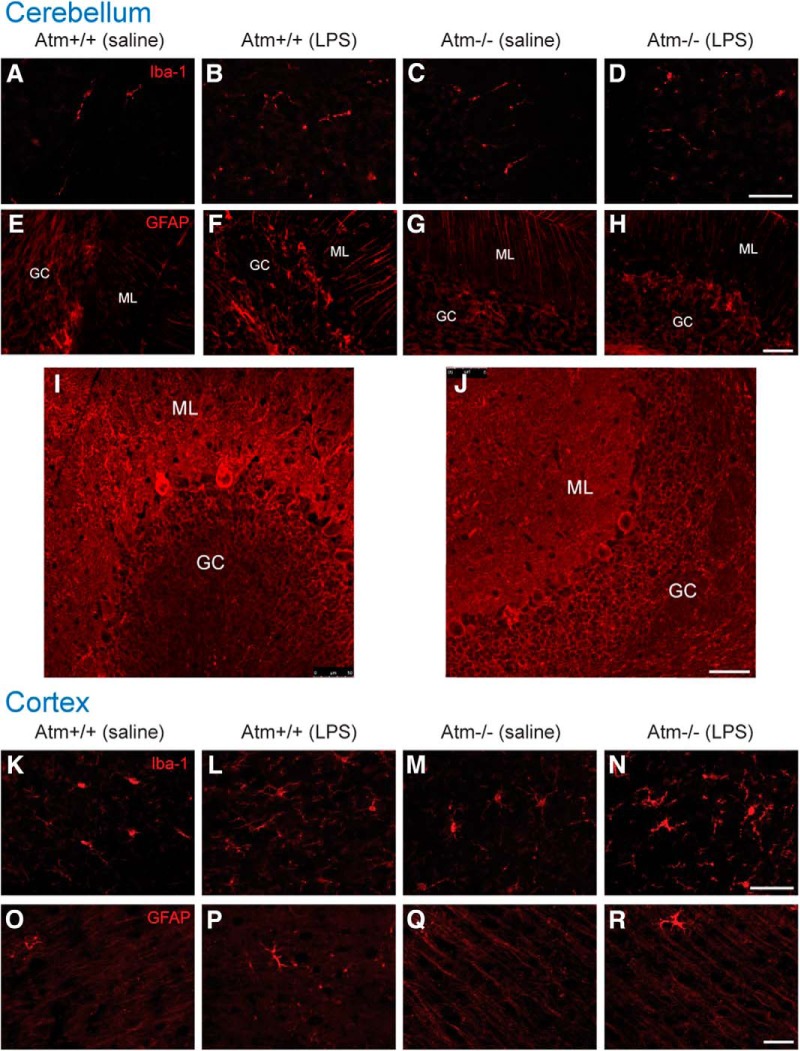
Ataxia-telangiectasia (A-T) is a multisystemic neurodegenerative disease of childhood caused by the absence of functional ATM (A-T mutated) protein. The cerebellar cortex has the most obvious neuropathology, yet cells in other brain regions are also abnormal. A-T mouse models have been produced that replicate much, though not all, of the complex A-T phenotype. Nongenetic factors, including modulations of the immune status of the animal, have also recently been found to play a role in the disease phenotype. Here we report that these modulations show both cytokine and brain region specificity. The CNS changes induced by broad-spectrum immune challenges, such as lipopolysaccharide (LPS) injections are a complex mixture of neuroprotective (TNFα) and neurodegenerative (IL1β) cytokine responses that change over time. For example, LPS first induces a protective response in A-T neurons through activation of tissue repair genes through infiltration of monocytes with M2 phenotype, followed over time by a set of more degenerative responses. Additional phenotypic complexity arises because the neuronal response to an immune challenge is regionally variable; cerebellum and cortex differ in important ways in their patterns of cellular and biochemical changes. Tracking these changes reveals an important though not exclusive role for the MAP kinase pathway. Our findings suggest brain responses to cytokine challenges are temporally and regionally specific and that both features are altered by the absence of ATM. This implies that management of the immune status of A-T patients might have significant clinical benefit.
Keywords: HDAC4; IL1beta; Purkinje cell; TNFalpha; ataxia-telangiectasia; cell cycle.
Conflict of interest statement
Authors report no conflicts of interest.
Figures
References
-
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21:383–421. - PMC - PubMed
-
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A (1996) Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86:159–171. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous