

cAMP-dependent Protein Kinase (PKA) Signaling Is Impaired in the Diabetic Heart
- PMID: 26468277
- PMCID: PMC4705931
- DOI: 10.1074/jbc.M115.681767
cAMP-dependent Protein Kinase (PKA) Signaling Is Impaired in the Diabetic Heart
Abstract
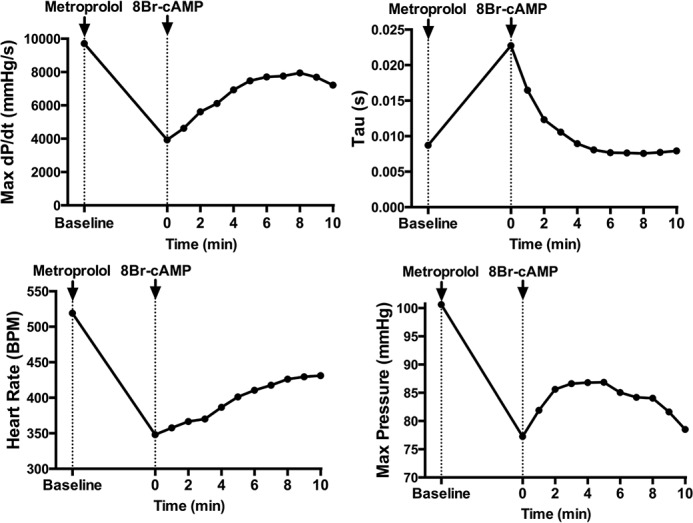
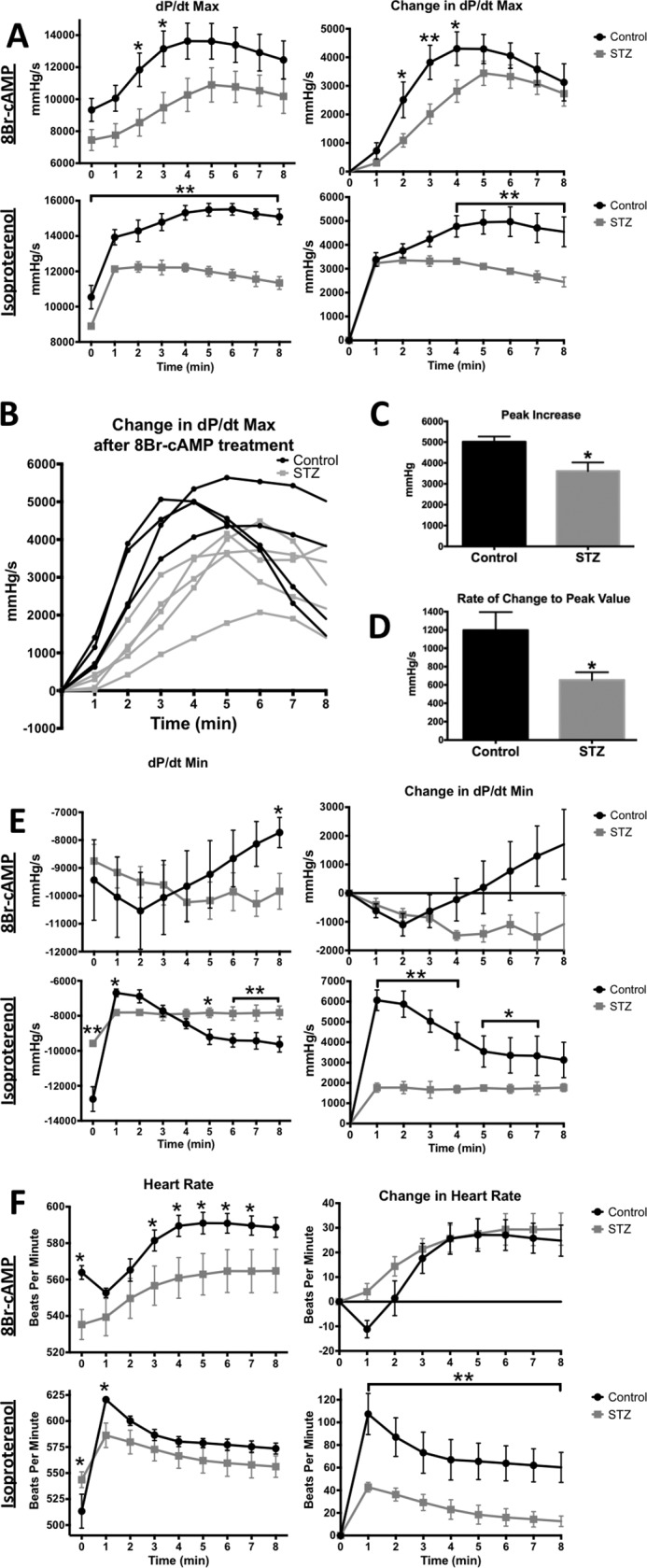
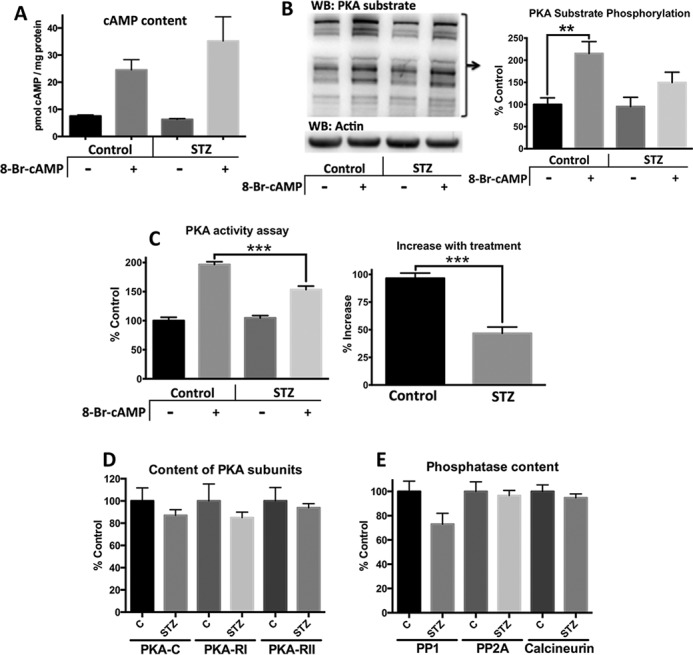
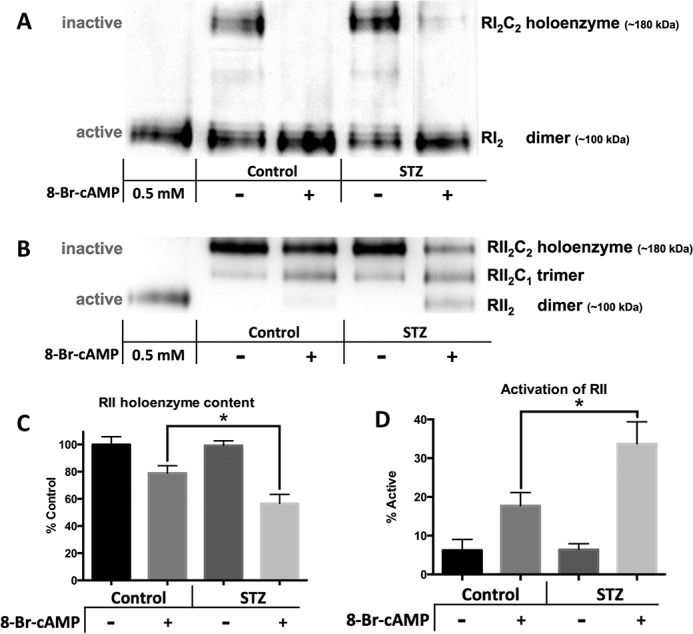
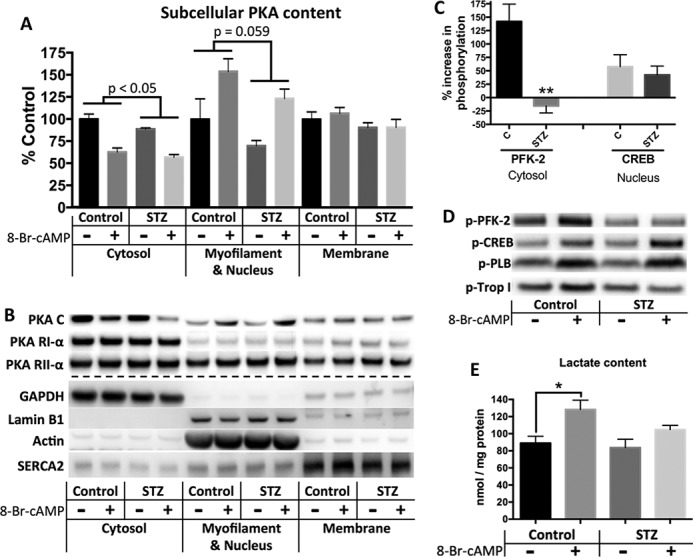
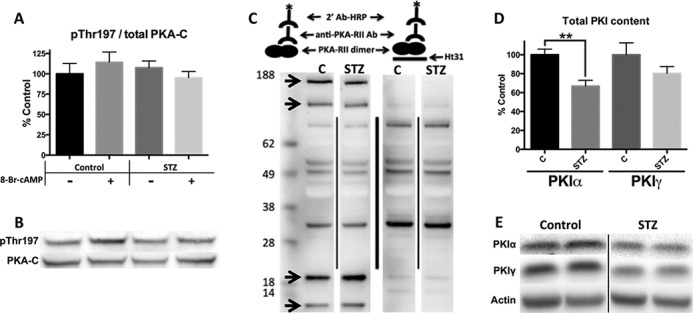
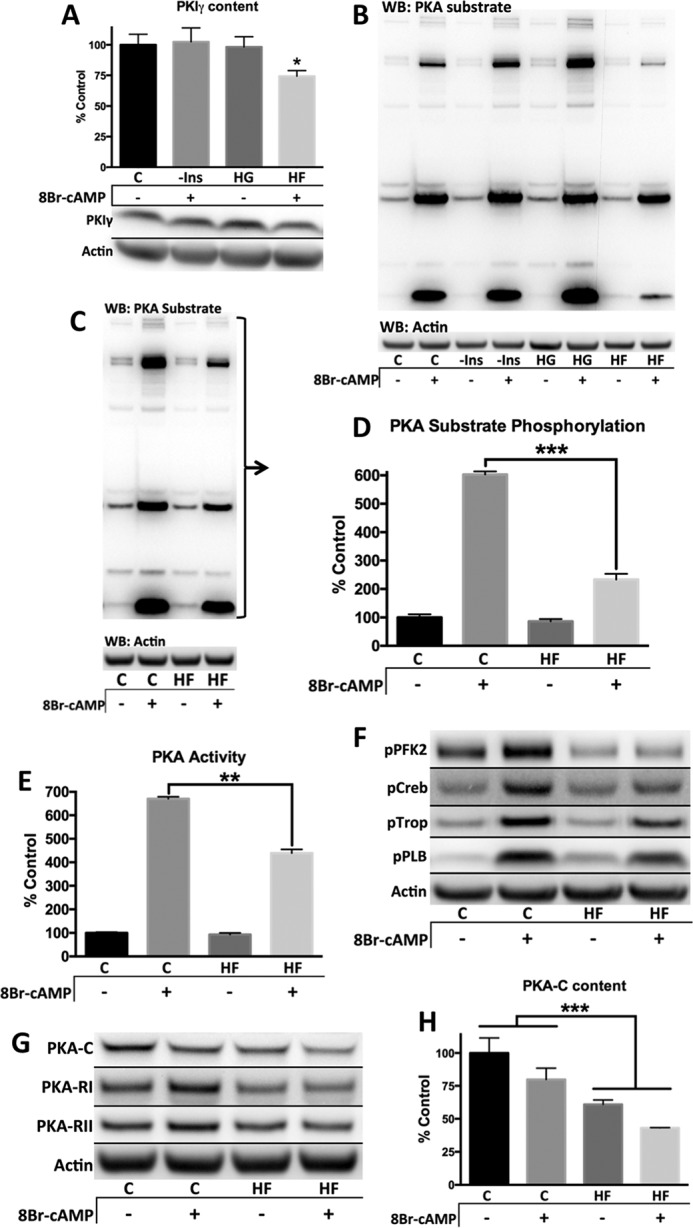
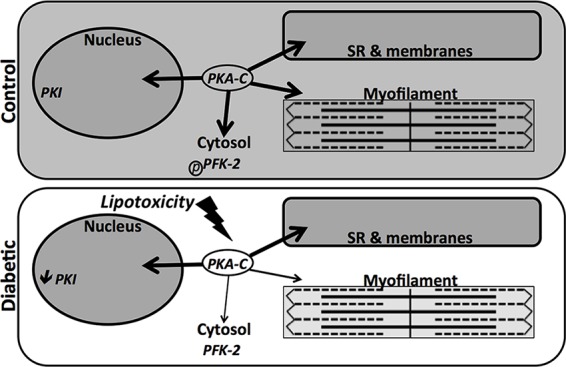
Diabetes mellitus causes cardiac dysfunction and heart failure that is associated with metabolic abnormalities and autonomic impairment. Autonomic control of ventricular function occurs through regulation of cAMP-dependent protein kinase (PKA). The diabetic heart has suppressed β-adrenergic responsiveness, partly attributable to receptor changes, yet little is known about how PKA signaling is directly affected. Control and streptozotocin-induced diabetic mice were therefore administered 8-bromo-cAMP (8Br-cAMP) acutely to activate PKA in a receptor-independent manner, and cardiac hemodynamic function and PKA signaling were evaluated. In response to 8Br-cAMP treatment, diabetic mice had impaired inotropic and lusitropic responses, thus demonstrating postreceptor defects. This impaired signaling was mediated by reduced PKA activity and PKA catalytic subunit content in the cytoplasm and myofilaments. Compartment-specific loss of PKA was reflected by reduced phosphorylation of discrete substrates. In response to 8Br-cAMP treatment, the glycolytic activator PFK-2 was robustly phosphorylated in control animals but not diabetics. Control adult cardiomyocytes cultured in lipid-supplemented media developed similar changes in PKA signaling, suggesting that lipotoxicity is a contributor to diabetes-induced β-adrenergic signaling dysfunction. This work demonstrates that PKA signaling is impaired in diabetes and suggests that treating hyperlipidemia is vital for proper cardiac signaling and function.
Keywords: cardiac metabolism; cardiomyocyte; diabetes; heart; protein kinase A (PKA).
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Gilbert R. E., and Krum H. (2015) Heart failure in diabetes: effects of anti-hyperglycaemic drug therapy. Lancet 385, 2107–2117 - PubMed
-
- Kannel W. B., Hjortland M., and Castelli W. P. (1974) Role of diabetes in congestive heart failure: the Framingham study. Am. J. Cardiol. 34, 29–34 - PubMed
-
- Aneja A., Tang W. H., Bansilal S., Garcia M. J., and Farkouh M. E. (2008) Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am. J. Med. 121, 748–757 - PubMed
-
- McMurray J. J., Gerstein H. C., Holman R. R., and Pfeffer M. A. (2014) Heart failure: a cardiovascular outcome in diabetes that can no longer be ignored. Lancet Diabetes Endocrinol. 2, 843–851 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
