

A novel immunofluorescent assay to investigate oxidative phosphorylation deficiency in mitochondrial myopathy: understanding mechanisms and improving diagnosis
- PMID: 26469001
- PMCID: PMC4606788
- DOI: 10.1038/srep15037
A novel immunofluorescent assay to investigate oxidative phosphorylation deficiency in mitochondrial myopathy: understanding mechanisms and improving diagnosis
Abstract
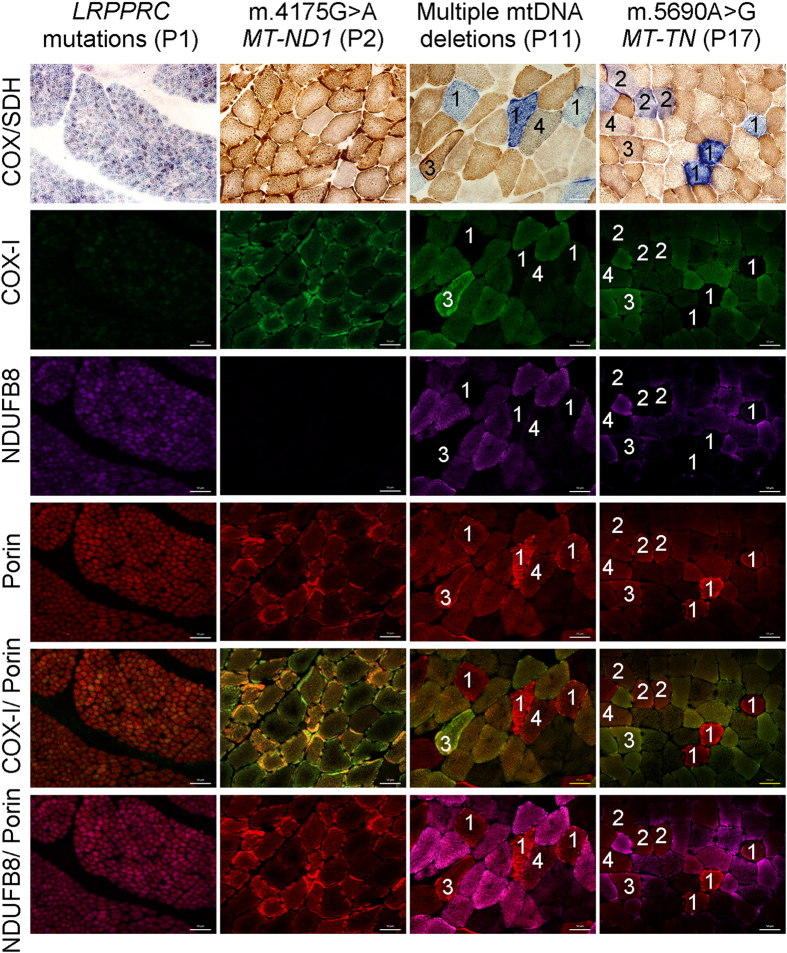
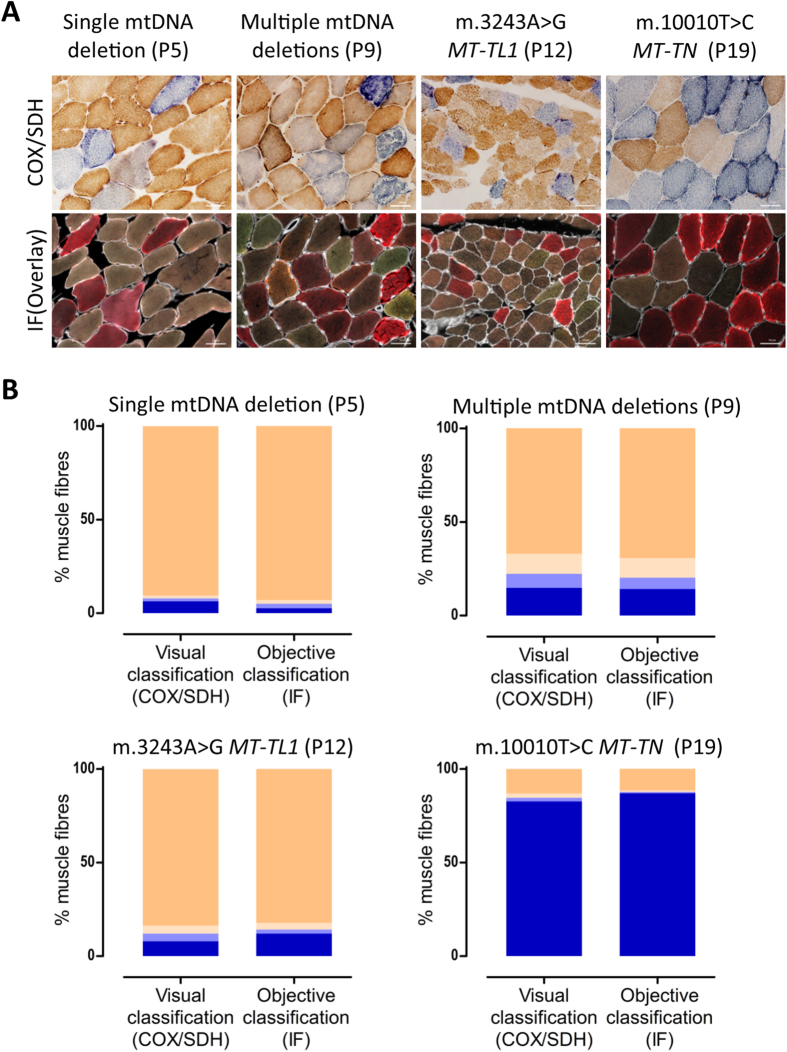
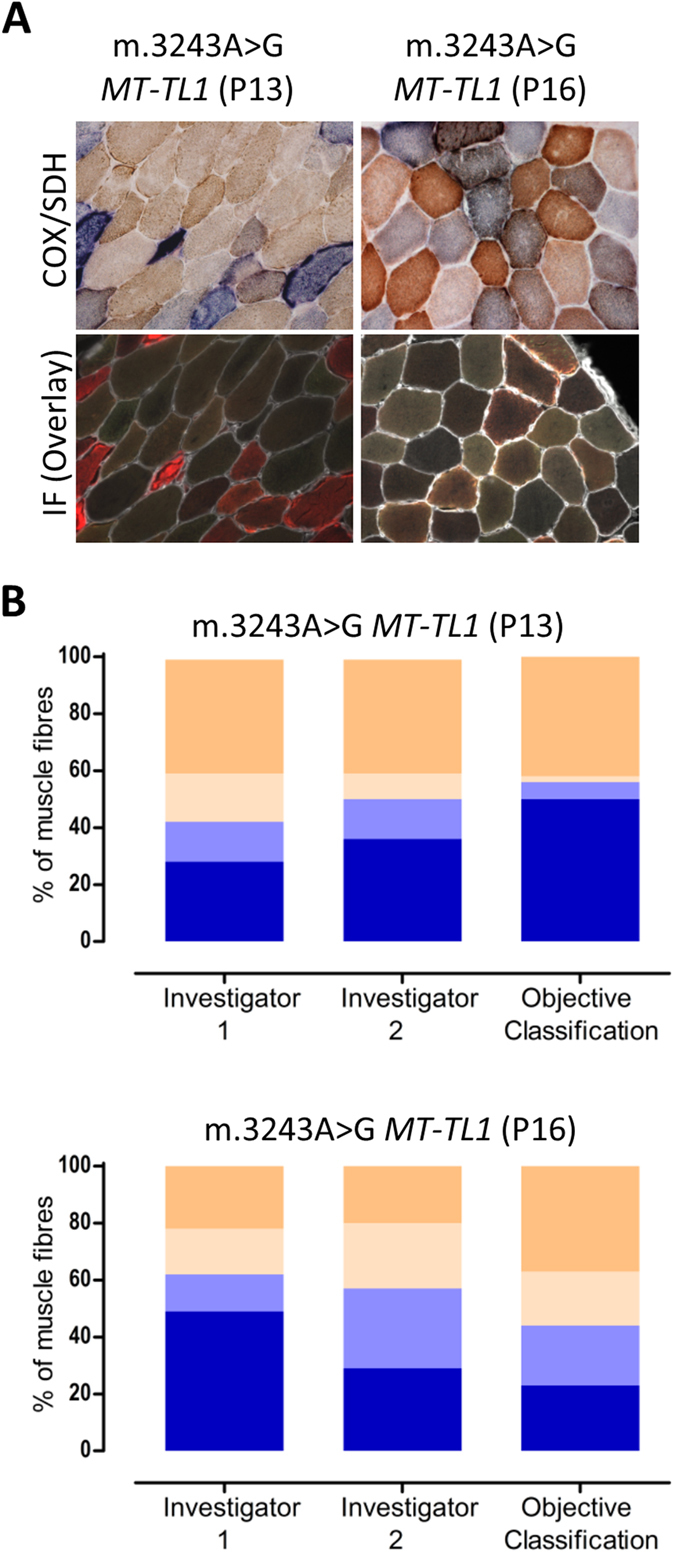
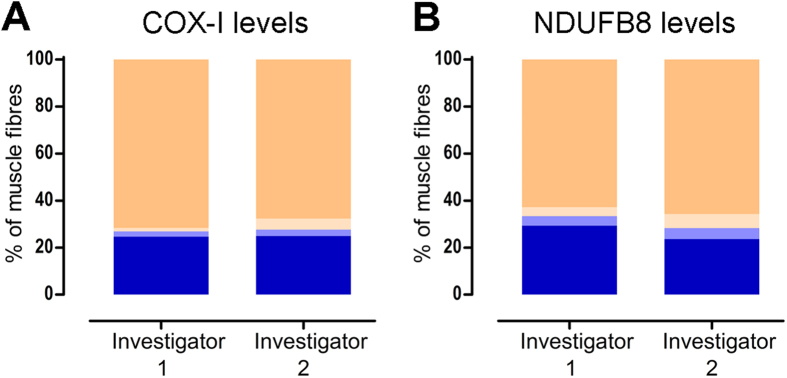
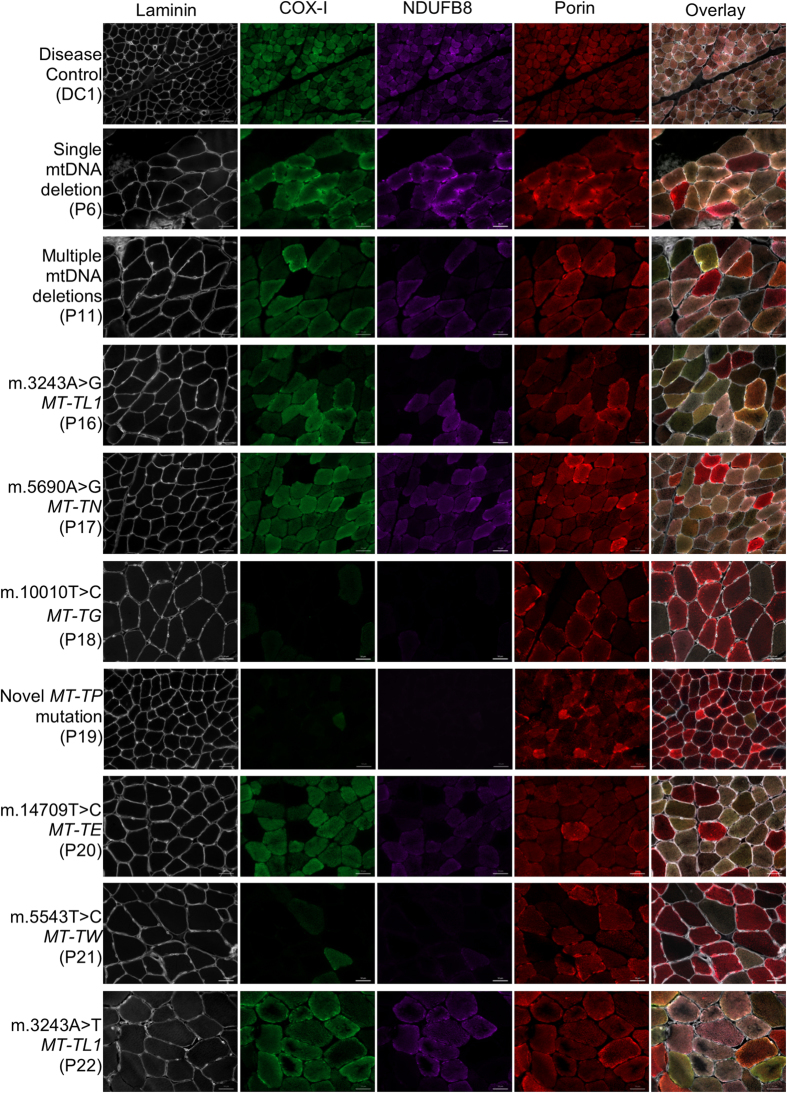
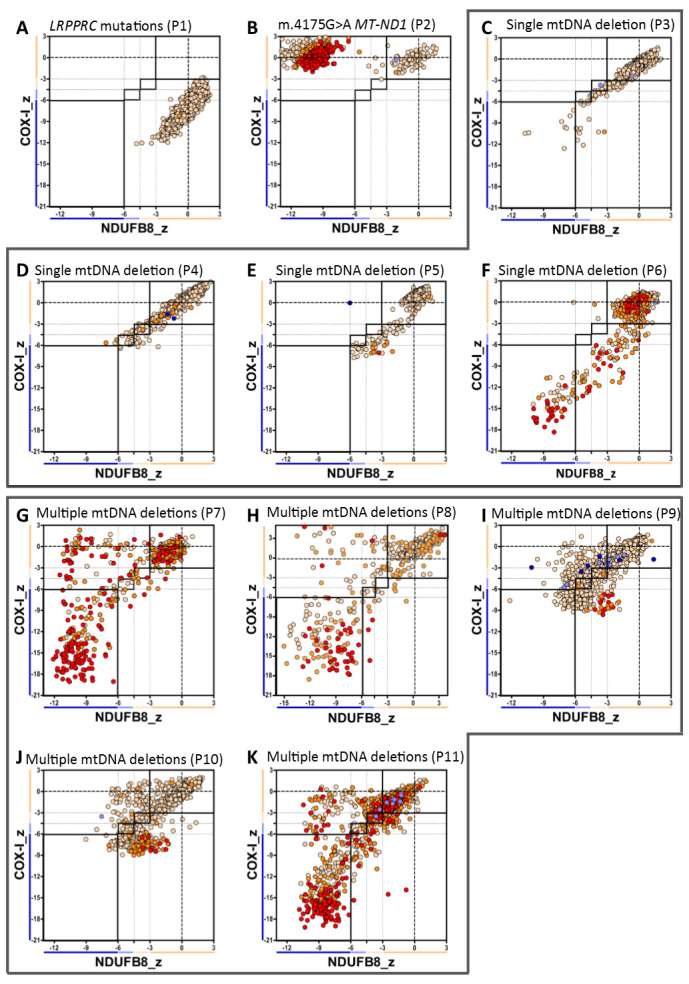
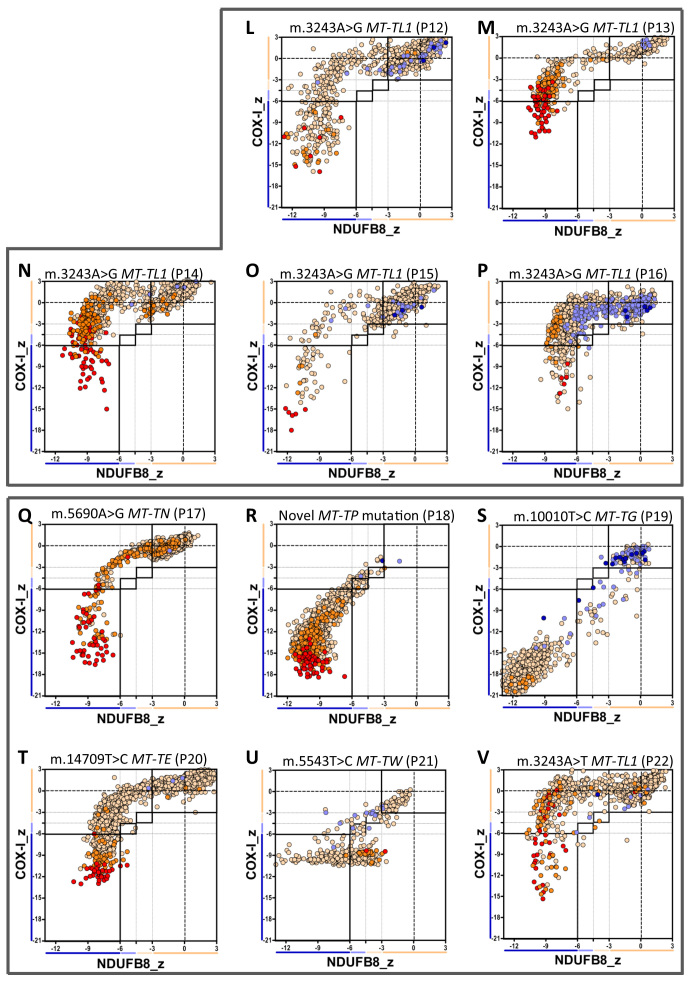
Oxidative phosphorylation defects in human tissues are often challenging to quantify due to a mosaic pattern of deficiency. Biochemical assays are difficult to interpret due to the varying enzyme deficiency levels found in individual cells. Histochemical analysis allows semi-quantitative assessment of complex II and complex IV activities, but there is no validated histochemical assay to assess complex I activity which is frequently affected in mitochondrial pathology. To help improve the diagnosis of mitochondrial disease and to study the mechanisms underlying mitochondrial abnormalities in disease, we have developed a quadruple immunofluorescent technique enabling the quantification of key respiratory chain subunits of complexes I and IV, together with an indicator of mitochondrial mass and a cell membrane marker. This assay gives precise and objective quantification of protein abundance in large numbers of individual muscle fibres. By assessing muscle biopsies from subjects with a range of different mitochondrial genetic defects we have demonstrated that specific genotypes exhibit distinct biochemical signatures in muscle, providing evidence for the diagnostic use of the technique, as well as insight into the underlying molecular pathology. Stringent testing for reproducibility and sensitivity confirms the potential value of the technique for mechanistic studies of disease and in the evaluation of therapeutic approaches.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
