

Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53
- PMID: 26471002
- PMCID: PMC5426523
- DOI: 10.1038/ncomms9651
Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53
Abstract
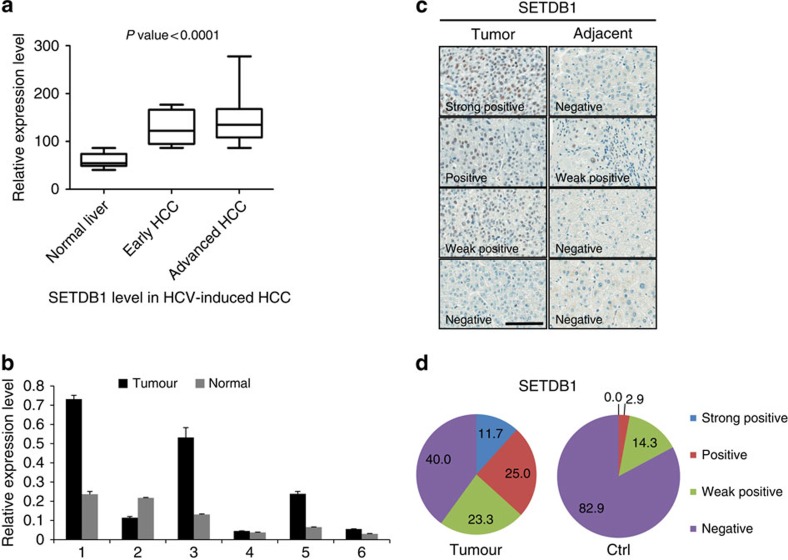
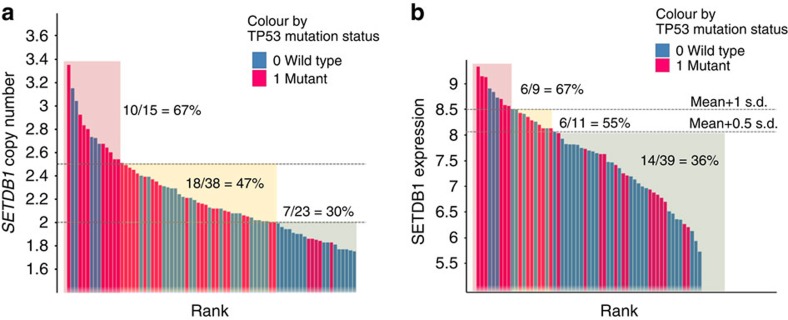
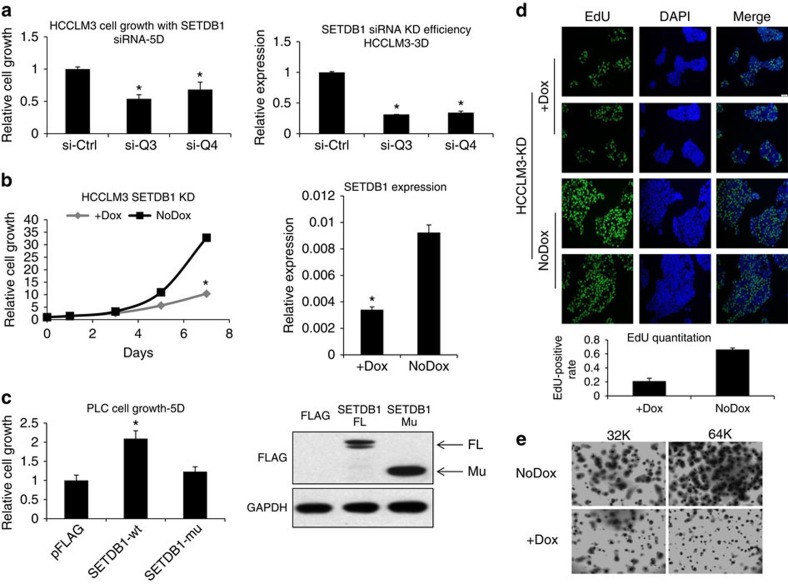
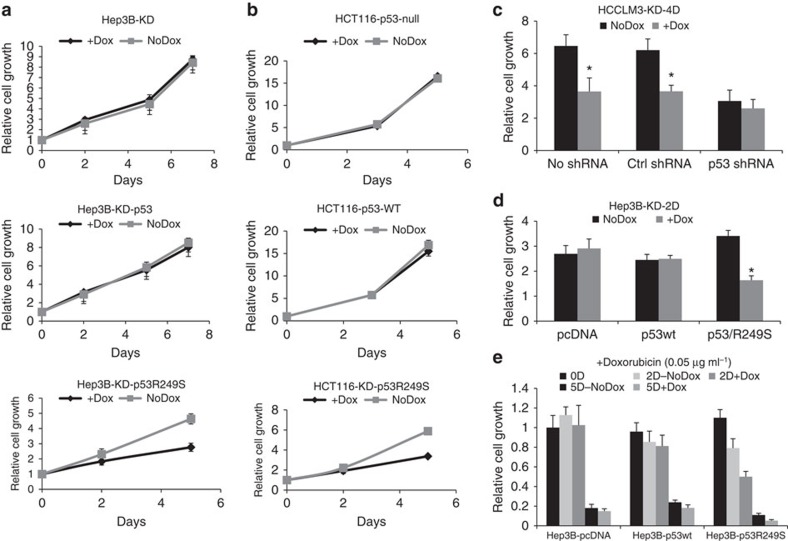
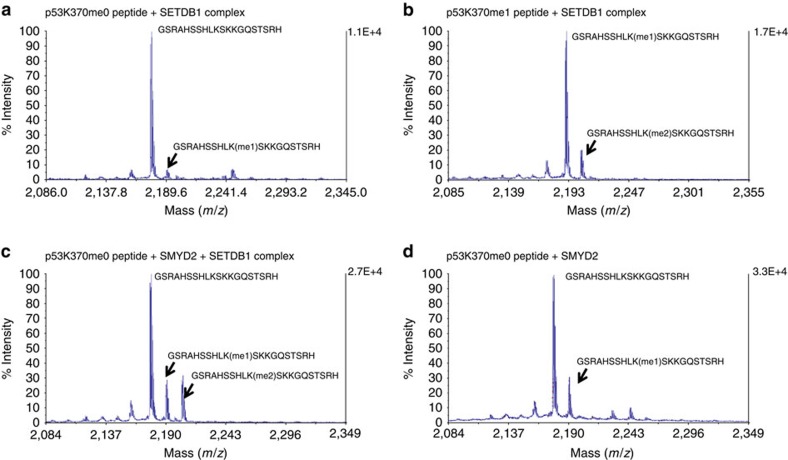
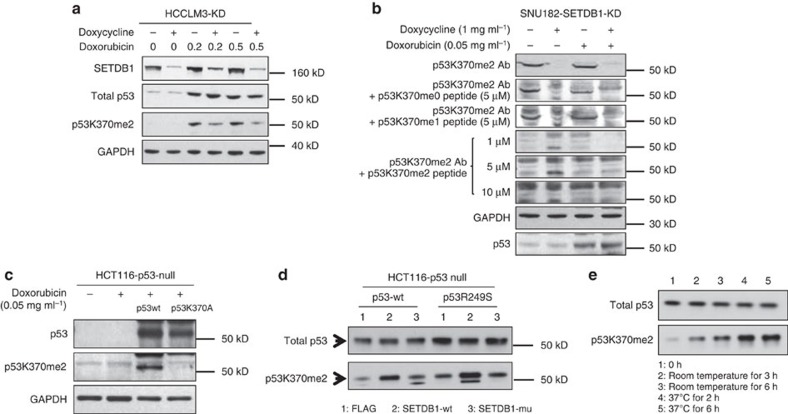
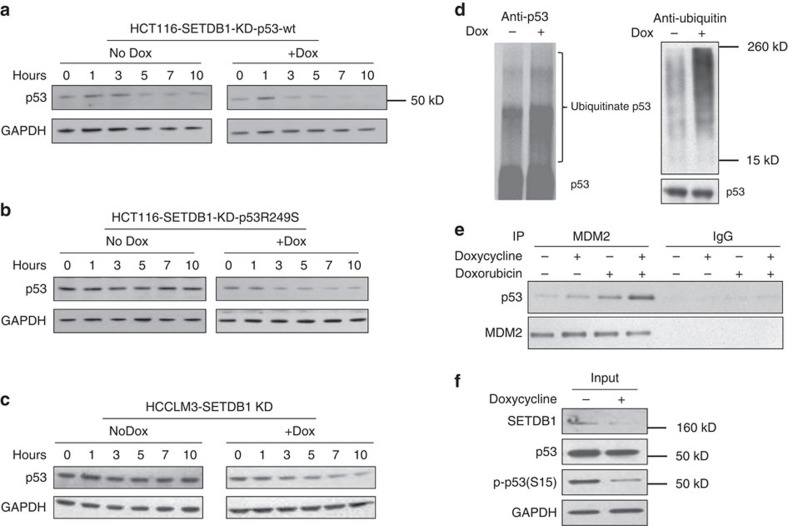
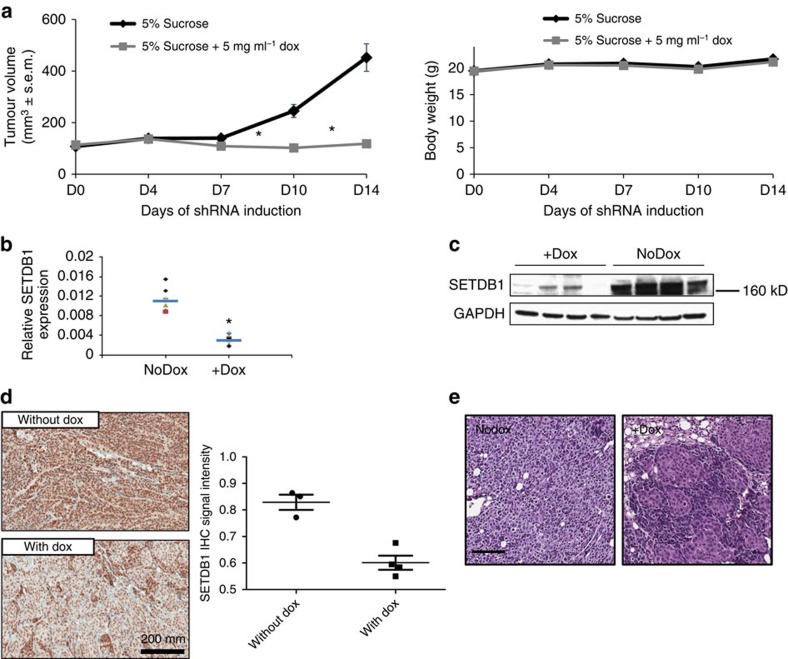
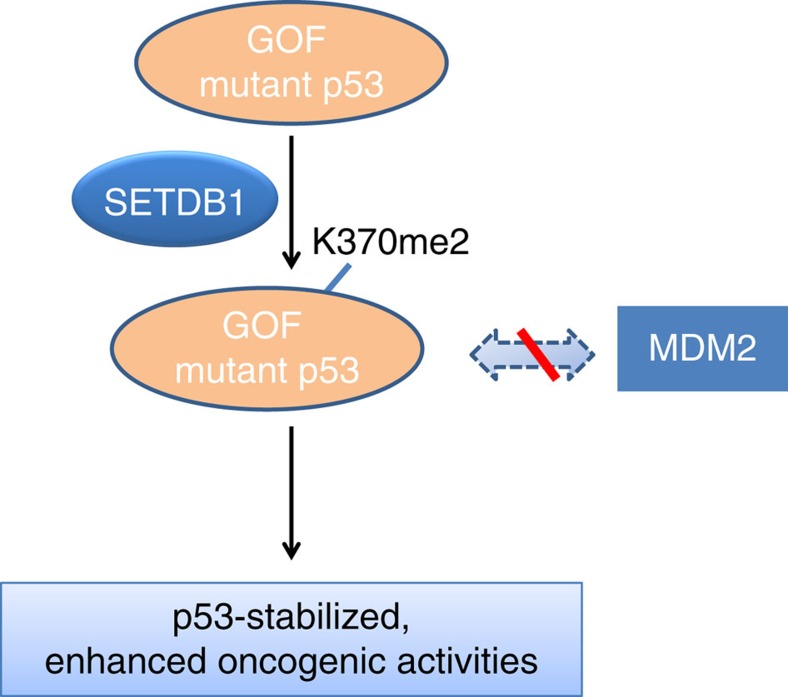
SETDB1 is a histone H3K9 methyltransferase that has a critical role in early development. It is located within a melanoma susceptibility locus and facilitates melanoma formation. However, the mechanism by which SETDB1 regulates tumorigenesis remains unknown. Here we report the molecular interplay between SETDB1 and the well-known hotspot gain-of-function (GOF) TP53 R249S mutation. We show that in hepatocellular carcinoma (HCC) SETDB1 is overexpressed with moderate copy number gain, and GOF TP53 mutations including R249S associate with this overexpression. Inactivation of SETDB1 in HCC cell lines bearing the R249S mutation suppresses cell growth. The TP53 mutation status renders cancer cells dependent on SETDB1. Moreover, SETDB1 forms a complex with p53 and catalyses p53K370 di-methylation. SETDB1 attenuation reduces the p53K370me2 level, which subsequently leads to increased recognition and degradation of p53 by MDM2. Together, we provide both genetic and biochemical evidence for a mechanism by which SETDB1 regulates cancer cell growth via methylation of p53.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Brosh R. & Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer 9, 701–713 (2009). - PubMed
-
- Shen H. M. & Ong C. N. Mutations of the p53 tumor suppressor gene and ras oncogenes in aflatoxin hepatocarcinogenesis. Mutat. Res. 366, 23–44 (1996). - PubMed
-
- Bressac B., Kew M., Wands J. & Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 350, 429–431 (1991). - PubMed
-
- Hsu I. C. et al.. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 350, 427–428 (1991). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
