

The PIM inhibitor AZD1208 synergizes with ruxolitinib to induce apoptosis of ruxolitinib sensitive and resistant JAK2-V617F-driven cells and inhibit colony formation of primary MPN cells
- PMID: 26472029
- PMCID: PMC4741885
- DOI: 10.18632/oncotarget.5653
The PIM inhibitor AZD1208 synergizes with ruxolitinib to induce apoptosis of ruxolitinib sensitive and resistant JAK2-V617F-driven cells and inhibit colony formation of primary MPN cells
Abstract
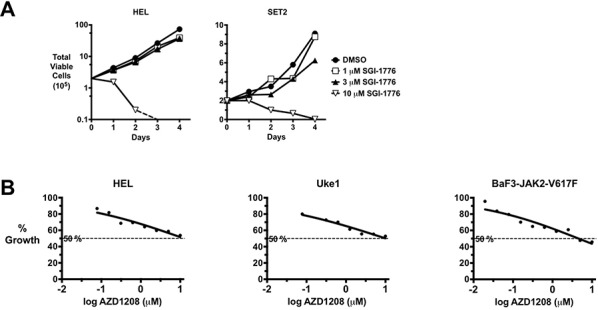
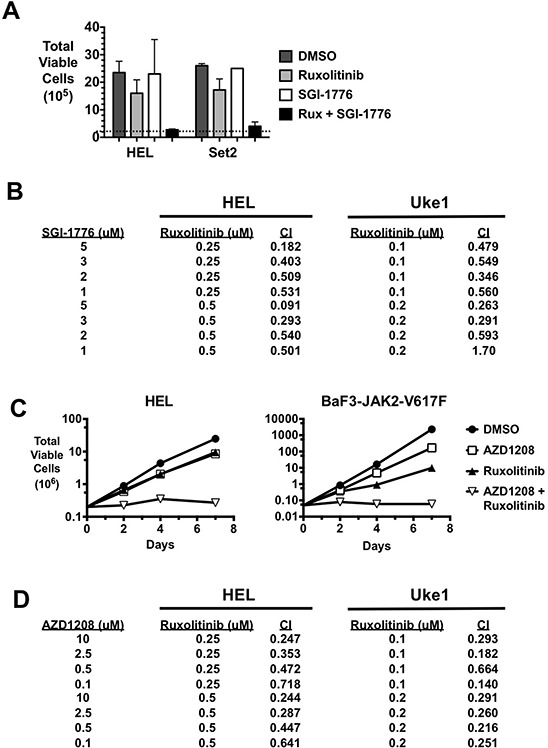
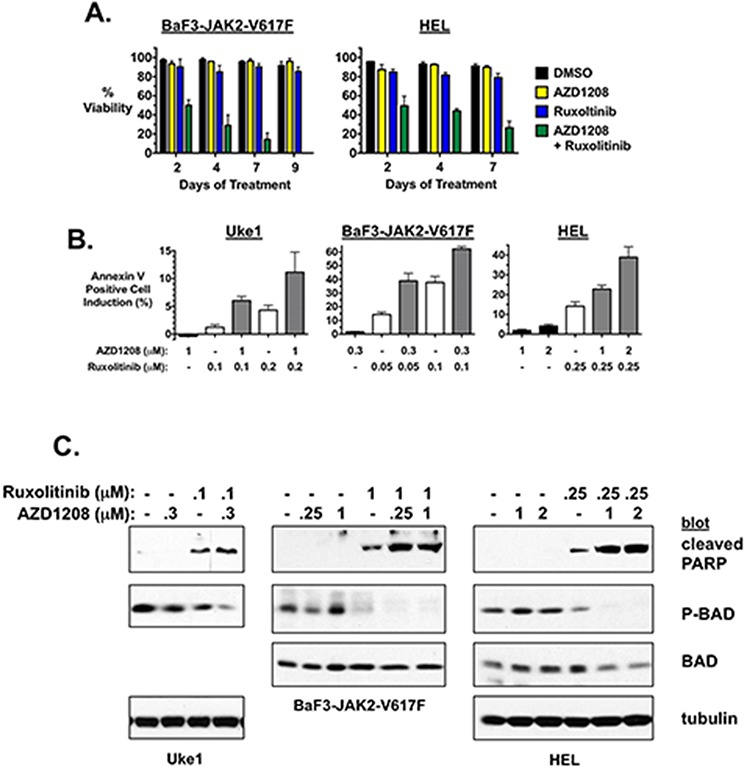
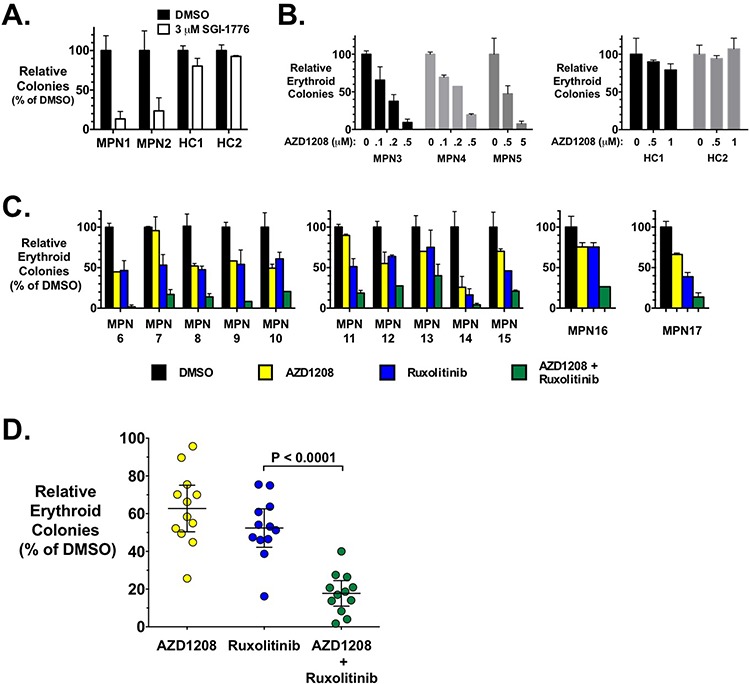
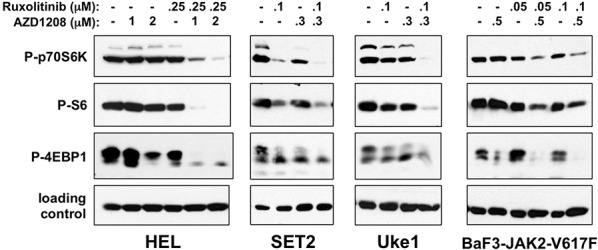
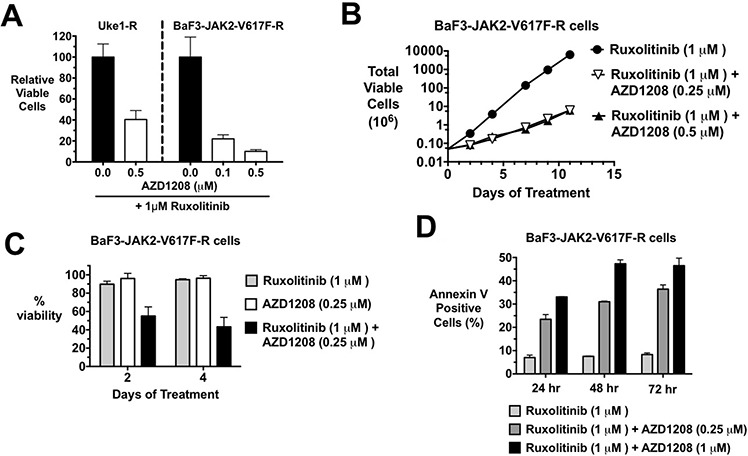
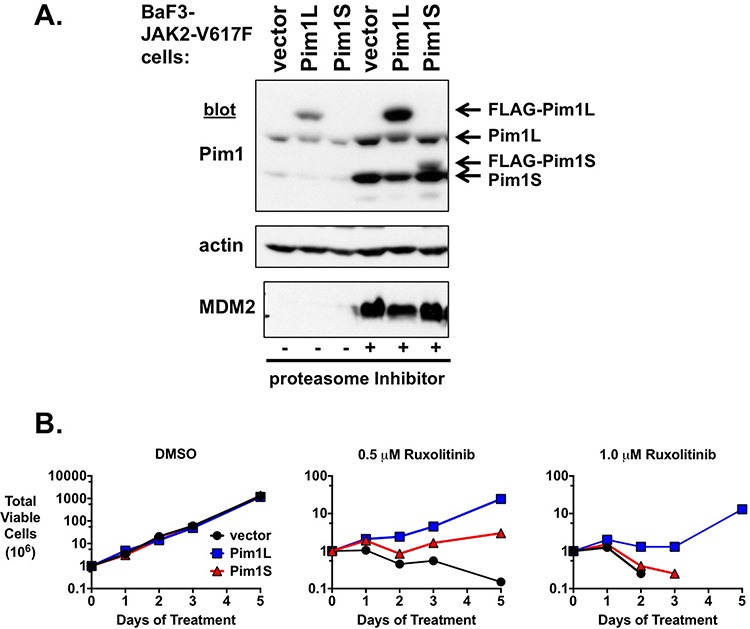
Classical myeloproliferative neoplasms (MPNs) are hematopoietic stem cell disorders that exhibit excess mature myeloid cells, bone marrow fibrosis, and risk of leukemic transformation. Aberrant JAK2 signaling plays an etiological role in MPN formation. Because neoplastic cells in patients are largely insensitive to current anti-JAK2 therapies, effective therapies remain needed. Members of the PIM family of serine/threonine kinases are induced by JAK/STAT signaling, regulate hematopoietic stem cell growth, protect hematopoietic cells from apoptosis, and exhibit hematopoietic cell transforming properties. We hypothesized that PIM kinases may offer a therapeutic target for MPNs. We treated JAK2-V617F-dependent MPN model cells as well as primary MPN patient cells with the PIM kinase inhibitors SGI-1776 and AZD1208 and the JAK2 inhibitor ruxolitinib. While MPN model cells were rather insensitive to PIM inhibitors, combination of PIM inhibitors with ruxolitinib led to a synergistic effect on MPN cell growth due to enhanced apoptosis. Importantly, PIM inhibitor mono-therapy inhibited, and AZD1208/ruxolitinib combination therapy synergistically suppressed, colony formation of primary MPN cells. Enhanced apoptosis by combination therapy was associated with activation of BAD, inhibition of downstream components of the mTOR pathway, including p70S6K and S6 protein, and activation of 4EBP1. Importantly, PIM inhibitors re-sensitized ruxolitinib-resistant MPN cells to ruxolitinib by inducing apoptosis. Finally, exogenous expression of PIM1 induced ruxolitinib resistance in MPN model cells. These data indicate that PIMs may play a role in MPNs and that combining PIM and JAK2 kinase inhibitors may offer a more efficacious therapeutic approach for MPNs over JAK2 inhibitor mono-therapy.
Keywords: JAK2-V617F; MPN; PIM; kinase inhibitor; therapy.
Conflict of interest statement
D. Huszar is an employee of AstraZeneca. All other authors declare no conflict of interest.
Figures
Similar articles
-
The pan-PIM inhibitor INCB053914 displays potent synergy in combination with ruxolitinib in models of MPN.Blood Adv. 2019 Nov 26;3(22):3503-3514. doi: 10.1182/bloodadvances.2019000260. Blood Adv. 2019. PMID: 31725895 Free PMC article.
-
mTOR inhibitors alone and in combination with JAK2 inhibitors effectively inhibit cells of myeloproliferative neoplasms.PLoS One. 2013;8(1):e54826. doi: 10.1371/journal.pone.0054826. Epub 2013 Jan 31. PLoS One. 2013. PMID: 23382981 Free PMC article.
-
Combination of PIM and JAK2 inhibitors synergistically suppresses MPN cell proliferation and overcomes drug resistance.Oncotarget. 2014 May 30;5(10):3362-74. doi: 10.18632/oncotarget.1951. Oncotarget. 2014. PMID: 24830942 Free PMC article.
-
Rationale for targeting the PI3K/Akt/mTOR pathway in myeloproliferative neoplasms.Clin Lymphoma Myeloma Leuk. 2013 Sep;13 Suppl 2:S307-9. doi: 10.1016/j.clml.2013.07.011. Clin Lymphoma Myeloma Leuk. 2013. PMID: 24290217 Review.
-
The Development and Use of Janus Kinase 2 Inhibitors for the Treatment of Myeloproliferative Neoplasms.Hematol Oncol Clin North Am. 2017 Aug;31(4):613-626. doi: 10.1016/j.hoc.2017.04.002. Epub 2017 May 17. Hematol Oncol Clin North Am. 2017. PMID: 28673391 Review.
Cited by
-
Targeting Abnormal Hematopoietic Stem Cells in Chronic Myeloid Leukemia and Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms.Int J Mol Sci. 2021 Jan 11;22(2):659. doi: 10.3390/ijms22020659. Int J Mol Sci. 2021. PMID: 33440869 Free PMC article. Review.
-
Bcl-xL represents a therapeutic target in Philadelphia negative myeloproliferative neoplasms.J Cell Mol Med. 2020 Sep;24(18):10978-10986. doi: 10.1111/jcmm.15730. Epub 2020 Aug 13. J Cell Mol Med. 2020. PMID: 32790151 Free PMC article.
-
Finding a Jill for JAK: Assessing Past, Present, and Future JAK Inhibitor Combination Approaches in Myelofibrosis.Cancers (Basel). 2020 Aug 14;12(8):2278. doi: 10.3390/cancers12082278. Cancers (Basel). 2020. PMID: 32823910 Free PMC article. Review.
-
Preclinical characterization of INCB053914, a novel pan-PIM kinase inhibitor, alone and in combination with anticancer agents, in models of hematologic malignancies.PLoS One. 2018 Jun 21;13(6):e0199108. doi: 10.1371/journal.pone.0199108. eCollection 2018. PLoS One. 2018. PMID: 29927999 Free PMC article.
-
Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms.Blood. 2017 Dec 28;130(26):2848-2859. doi: 10.1182/blood-2017-05-784942. Epub 2017 Oct 17. Blood. 2017. PMID: 29042365 Free PMC article.
References
-
- Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6:372–375. - PubMed
-
- Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011;29:573–582. - PubMed
-
- Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–683. - PubMed
-
- Geyer HL, Mesa RA. Therapy for myeloproliferative neoplasms: when, which agent, and how? Blood. 2014;124:3529–3537. - PubMed
-
- Levine RL, Wernig G. Role of JAK-STAT signaling in the pathogenesis of myeloproliferative disorders. Hematology Am Soc Hematol Educ Program. 2006;510:233–239. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
