

Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias
- PMID: 26475296
- PMCID: PMC4845125
- DOI: 10.1161/JAHA.115.002149
Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias
Abstract
Background: Patients with dilated cardiomyopathy (DCM) may present with ventricular arrhythmias early in the disease course, unrelated to the severity of left ventricular dysfunction. These patients may be classified as having an arrhythmogenic DCM (AR-DCM). We investigated the phenotype and natural history of patients with AR-DCM.
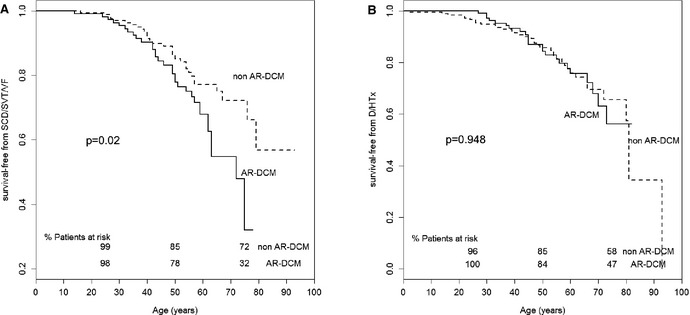
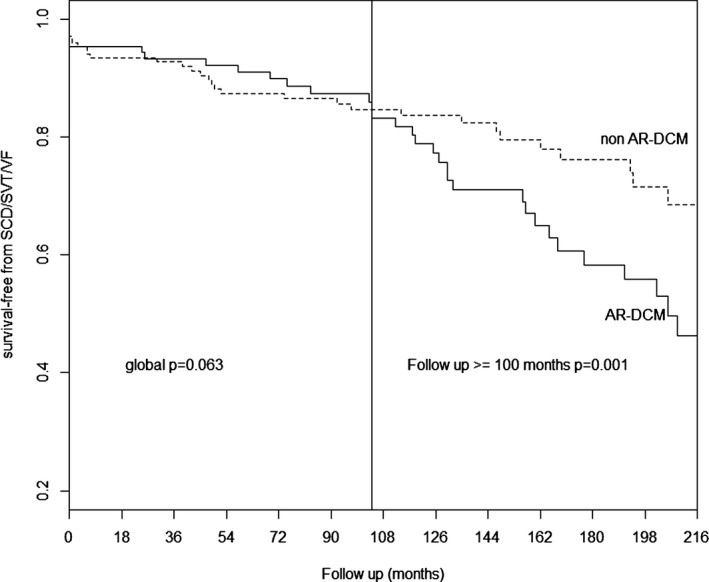
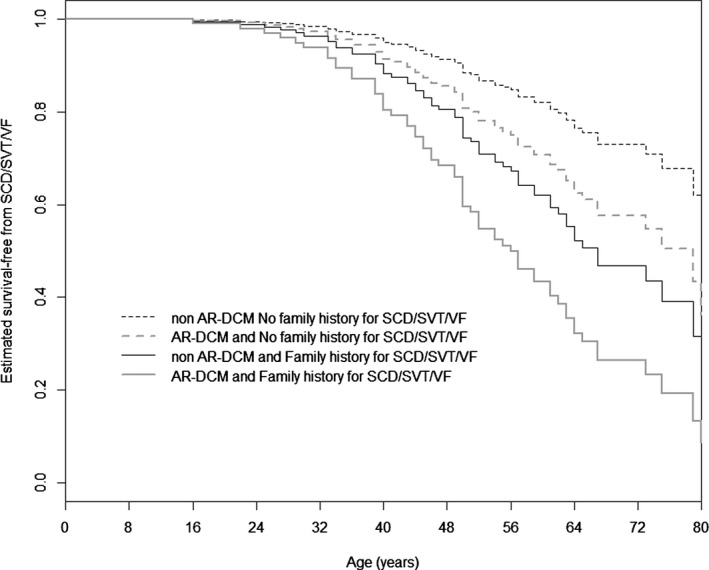
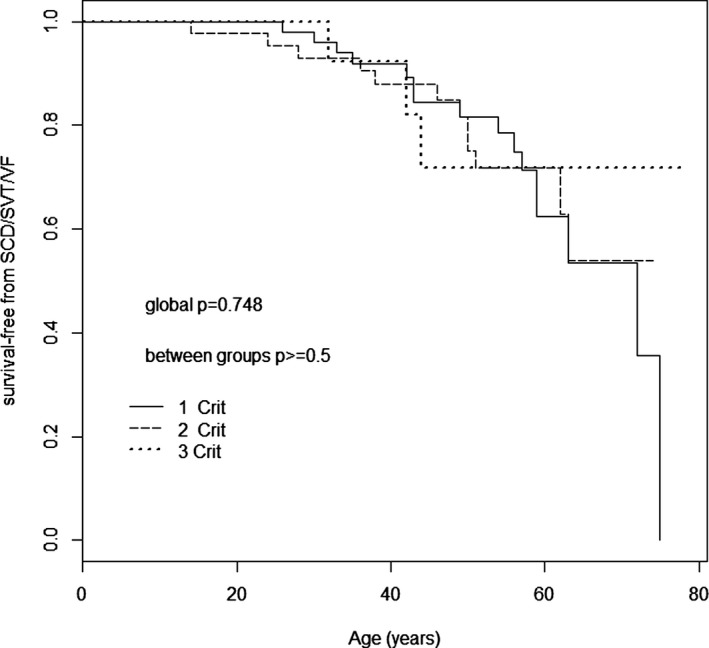
Methods and results: Two hundred eighty-five patients with a recent diagnosis of DCM (median duration of the disease 1 month, range 0 to 7 months) and who had Holter monitoring at baseline were comprehensively evaluated and followed for 107 months (range 29 to 170 months). AR-DCM was defined by the presence of ≥1 of the following: unexplained syncope, rapid nonsustained ventricular tachycardia (≥5 beats, ≥150 bpm), ≥1000 premature ventricular contractions/24 hours, and ≥50 ventricular couplets/24 hours, in the absence of overt heart failure. The primary end points were sudden cardiac death (SCD), sustained ventricular tachycardia (SVT), or ventricular fibrillation (VF). The secondary end points were death from congestive heart failure or heart transplantation. Of the 285 patients, 109 (38.2%) met criteria for AR-DCM phenotype. AR-DCM subjects had a higher incidence of SCD/SVT/VF compared with non-AR-DCM patients (30.3% vs 17.6%, P=0.022), with no difference in the secondary end points. A family history of SCD/SVT/VF and the AR-DCM phenotype were statistically significant and cumulative predictors of SCD/SVT/VF.
Conclusions: One-third of DCM patients may have an arrhythmogenic phenotype associated with increased risk of arrhythmias during follow-up. A family history of ventricular arrhythmias in DCM predicts a poor prognosis and increased risk of SCD.
Keywords: arrhythmia; cardiomyopathy; prognosis; sudden death.
© 2015 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley Blackwell.
Figures
References
-
- Sen‐Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left‐dominant arrhythmogenic cardiomyopathy: an under‐recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–2187. - PubMed
-
- Sen‐Chowdhry S, Syrris P, Pantazis A, Quarta G, McKenna WJ, Chambers JC. Mutational heterogeneity, modifier genes, and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ Cardiovasc Genet. 2010;3:323–330. - PubMed
-
- Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, Mahon NG, McKenna WJ. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212–2218. - PubMed
-
- Ritter M, Oechslin E, Sutsch G, Attenhofer C, Schneider J, Jenni R. Isolated noncompaction of the myocardium in adults. Mayo Clin Proc. 1997;72:26–31. - PubMed
-
- Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990;82:507–513. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
