

Prioritization of candidate genes in "QTL-hotspot" region for drought tolerance in chickpea (Cicer arietinum L.)
- PMID: 26478518
- PMCID: PMC4609953
- DOI: 10.1038/srep15296
Prioritization of candidate genes in "QTL-hotspot" region for drought tolerance in chickpea (Cicer arietinum L.)
Abstract
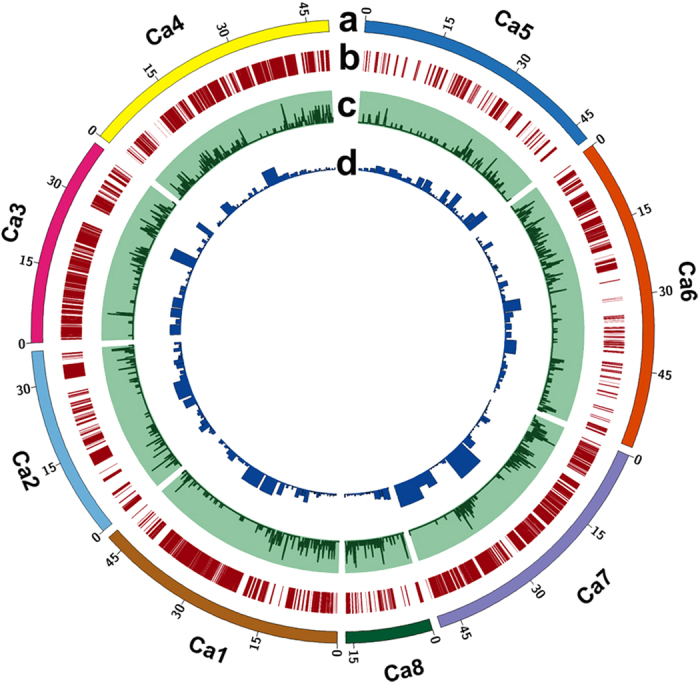
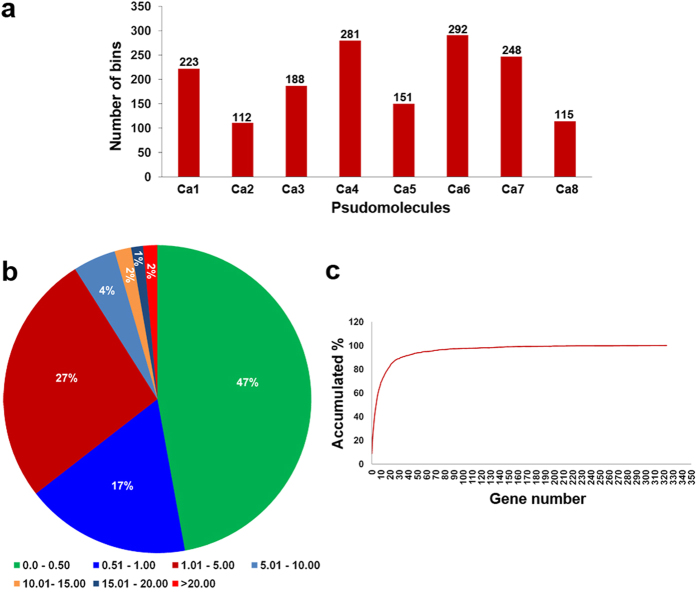
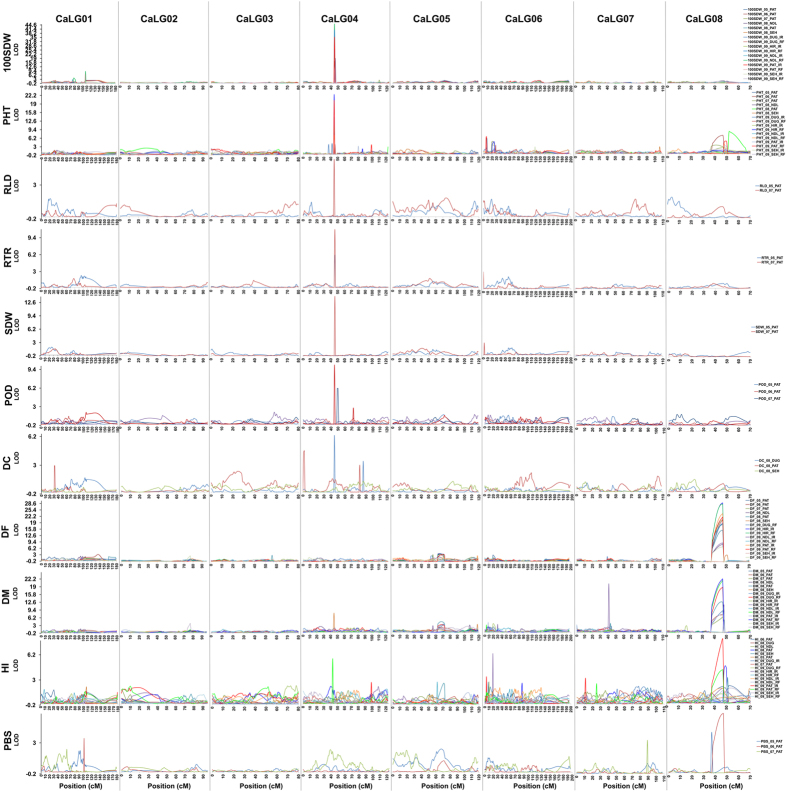
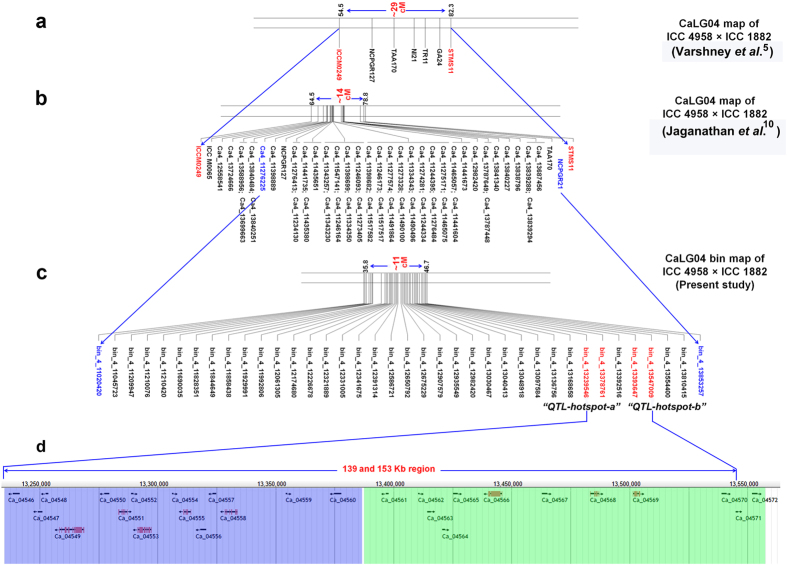
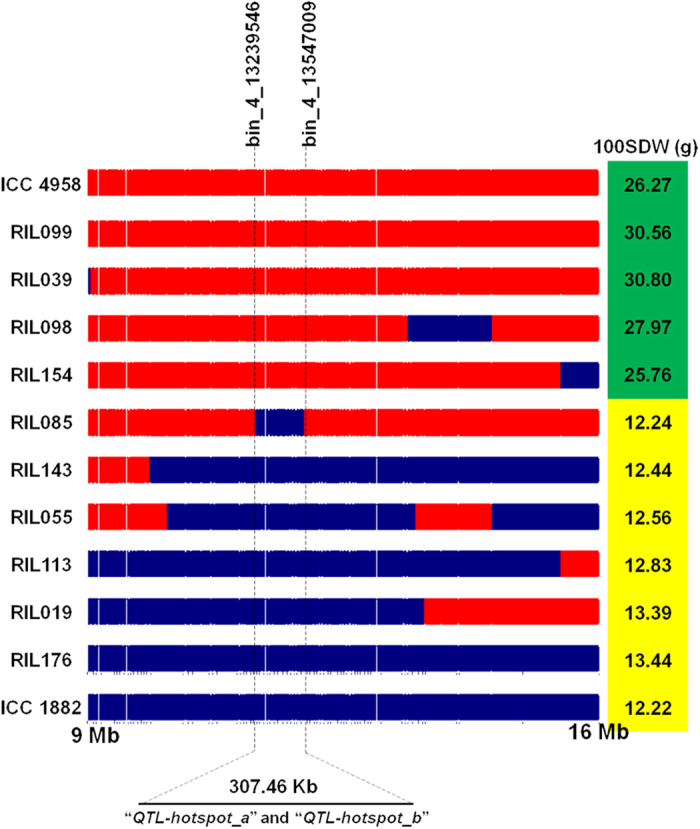
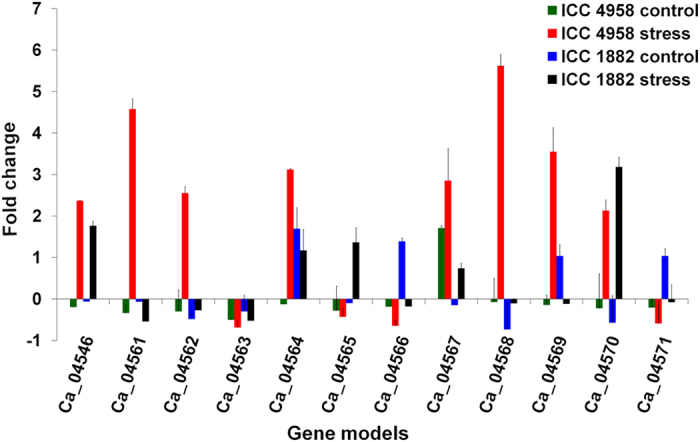
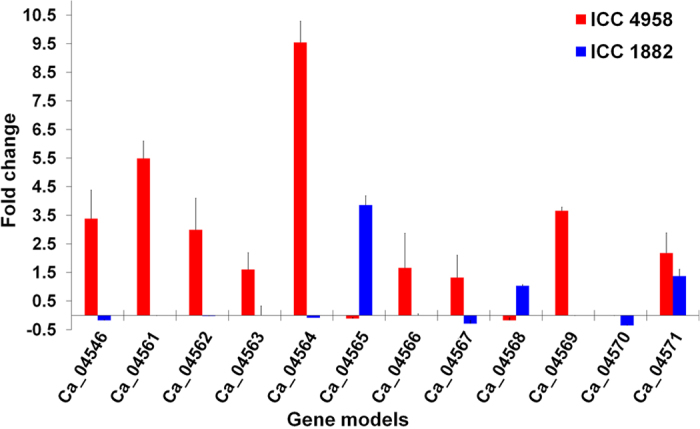
A combination of two approaches, namely QTL analysis and gene enrichment analysis were used to identify candidate genes in the "QTL-hotspot" region for drought tolerance present on the Ca4 pseudomolecule in chickpea. In the first approach, a high-density bin map was developed using 53,223 single nucleotide polymorphisms (SNPs) identified in the recombinant inbred line (RIL) population of ICC 4958 (drought tolerant) and ICC 1882 (drought sensitive) cross. QTL analysis using recombination bins as markers along with the phenotyping data for 17 drought tolerance related traits obtained over 1-5 seasons and 1-5 locations split the "QTL-hotspot" region into two subregions namely "QTL-hotspot_a" (15 genes) and "QTL-hotspot_b" (11 genes). In the second approach, gene enrichment analysis using significant marker trait associations based on SNPs from the Ca4 pseudomolecule with the above mentioned phenotyping data, and the candidate genes from the refined "QTL-hotspot" region showed enrichment for 23 genes. Twelve genes were found common in both approaches. Functional validation using quantitative real-time PCR (qRT-PCR) indicated four promising candidate genes having functional implications on the effect of "QTL-hotspot" for drought tolerance in chickpea.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

Similar articles
-
Genotyping-by-sequencing based intra-specific genetic map refines a ''QTL-hotspot" region for drought tolerance in chickpea.Mol Genet Genomics. 2015 Apr;290(2):559-71. doi: 10.1007/s00438-014-0932-3. Epub 2014 Oct 25. Mol Genet Genomics. 2015. PMID: 25344290 Free PMC article.
-
Plant vigour QTLs co-map with an earlier reported QTL hotspot for drought tolerance while water saving QTLs map in other regions of the chickpea genome.BMC Plant Biol. 2018 Feb 6;18(1):29. doi: 10.1186/s12870-018-1245-1. BMC Plant Biol. 2018. PMID: 29409451 Free PMC article.
-
Two key genomic regions harbour QTLs for salinity tolerance in ICCV 2 × JG 11 derived chickpea (Cicer arietinum L.) recombinant inbred lines.BMC Plant Biol. 2015 May 22;15:124. doi: 10.1186/s12870-015-0491-8. BMC Plant Biol. 2015. PMID: 25994494 Free PMC article.
-
Advances in genetics and molecular breeding of three legume crops of semi-arid tropics using next-generation sequencing and high-throughput genotyping technologies.J Biosci. 2012 Nov;37(5):811-20. doi: 10.1007/s12038-012-9228-0. J Biosci. 2012. PMID: 23107917 Review.
-
Using genetic mapping and genomics approaches in understanding and improving drought tolerance in pearl millet.J Exp Bot. 2011 Jan;62(2):397-408. doi: 10.1093/jxb/erq265. Epub 2010 Sep 5. J Exp Bot. 2011. PMID: 20819788 Review.
Cited by
-
A high-density SNP-based linkage map using genotyping-by-sequencing and its utilization for improved genome assembly of chickpea (Cicer arietinum L.).Funct Integr Genomics. 2020 Nov;20(6):763-773. doi: 10.1007/s10142-020-00751-y. Epub 2020 Aug 27. Funct Integr Genomics. 2020. PMID: 32856221
-
Transcriptome landscape of perennial wild Cicer microphyllum uncovers functionally relevant molecular tags regulating agronomic traits in chickpea.Sci Rep. 2016 Sep 29;6:33616. doi: 10.1038/srep33616. Sci Rep. 2016. PMID: 27680662 Free PMC article.
-
Genomic and phenotypic analysis of Vavilov's historic landraces reveals the impact of environment and genomic islands of agronomic traits.Sci Rep. 2017 Jul 6;7(1):4816. doi: 10.1038/s41598-017-05087-5. Sci Rep. 2017. PMID: 28684880 Free PMC article.
-
Identification of stable heat tolerance QTLs using inter-specific recombinant inbred line population derived from GPF 2 and ILWC 292.PLoS One. 2021 Aug 9;16(8):e0254957. doi: 10.1371/journal.pone.0254957. eCollection 2021. PLoS One. 2021. PMID: 34370732 Free PMC article.
-
Transgenic chickpea (Cicer arietinum L.) harbouring AtDREB1a are physiologically better adapted to water deficit.BMC Plant Biol. 2021 Jan 11;21(1):39. doi: 10.1186/s12870-020-02815-4. BMC Plant Biol. 2021. PMID: 33430800 Free PMC article.
References
-
- Jukanti A. K., Gaur P. M., Gowda C. L. L. & Chibbar R. N. Nutritional quality and health benefits of chickpea (Cicer arietinum L.): a review. Brit. J. Nutr. 108, S11–S26 (2012). - PubMed
-
- Ahmad F., Gaur P. M. & Croser J. Chickpea (Cicer arietinum L.). In: Genetic Resources, Chromosome Engineering, and Crop Improvement—Grain Legumes (eds Singh R. J., Jauhar P. P.). CRC Press (2005).
-
- Hamwieh A., Imtiaz M. & Malhotra R. S. Multi-environment QTL analyses for drought-related traits in a recombinant inbred population of chickpea (Cicer arientinum L.). Theor. Appl. Genet. 126, 1025–1038 (2013). - PubMed
-
- Rehman A. U. et al. Mapping QTL associated with traits affecting grain yield in chickpea (Cicer arietinum L.) under terminal drought stress. Crop Sci. 51, 450–463 (2011).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
