

Selector function of MHC I molecules is determined by protein plasticity
- PMID: 26482009
- PMCID: PMC5224517
- DOI: 10.1038/srep14928
Selector function of MHC I molecules is determined by protein plasticity
Abstract
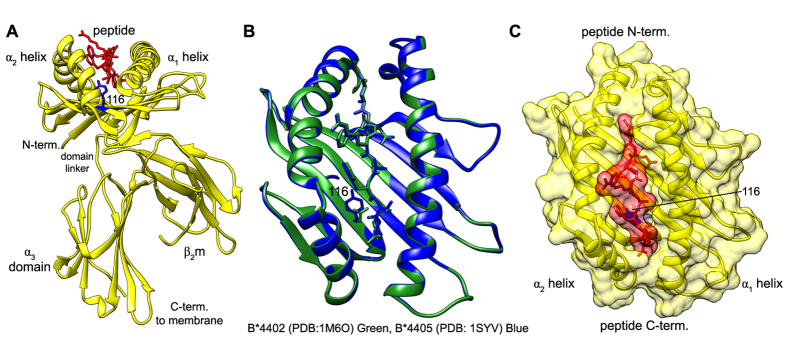
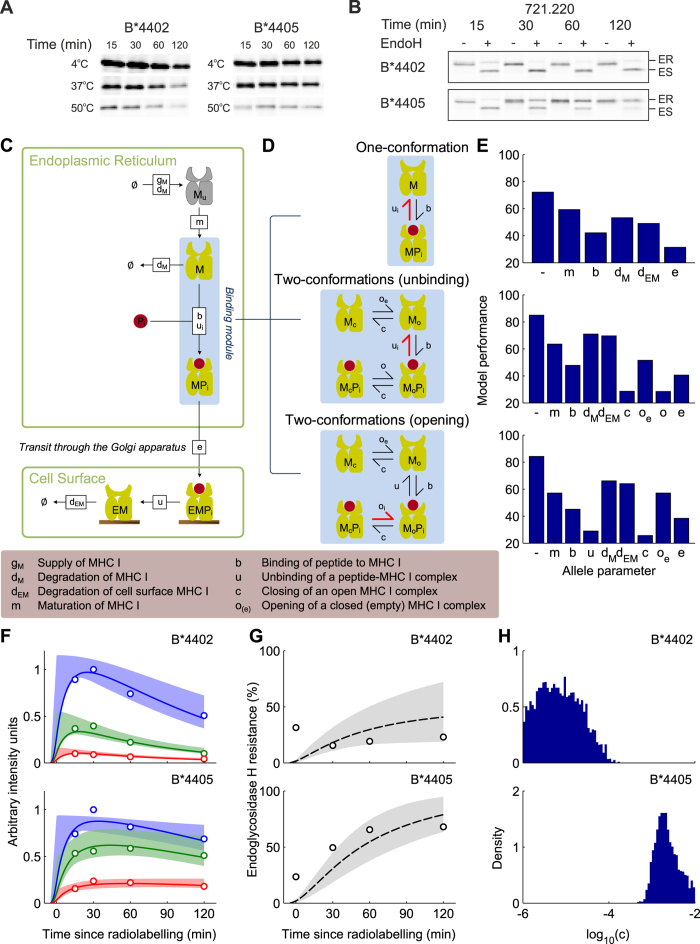
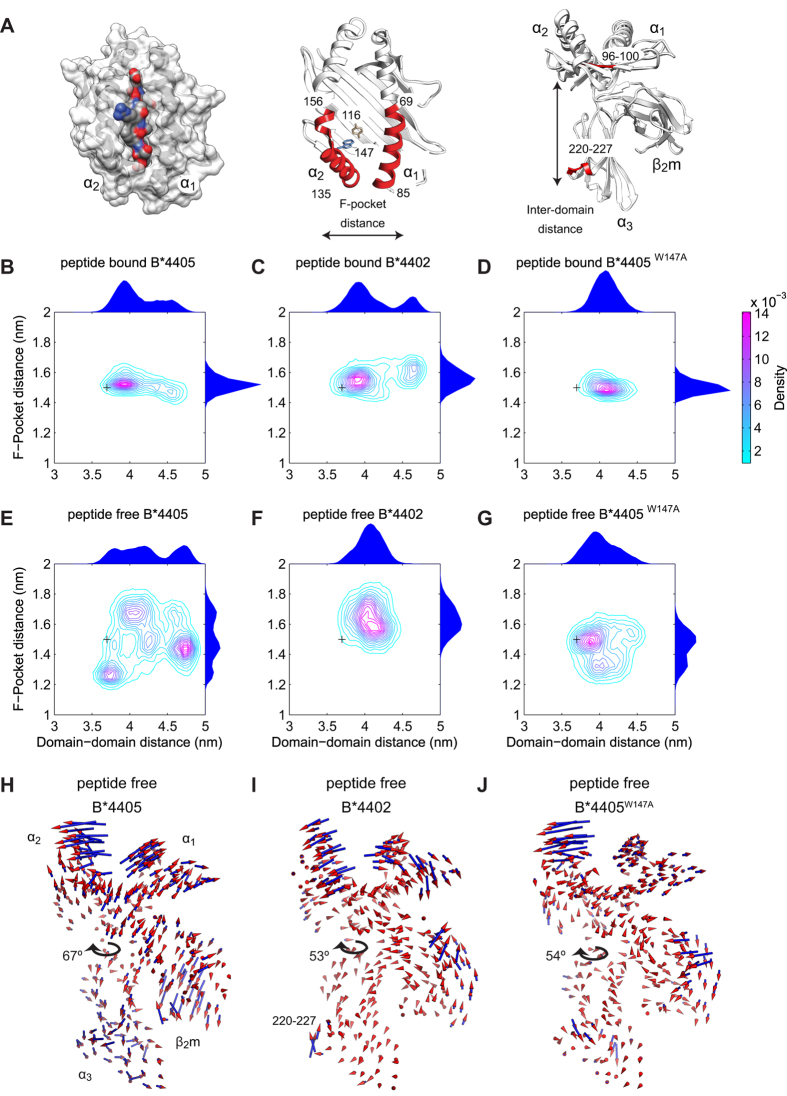
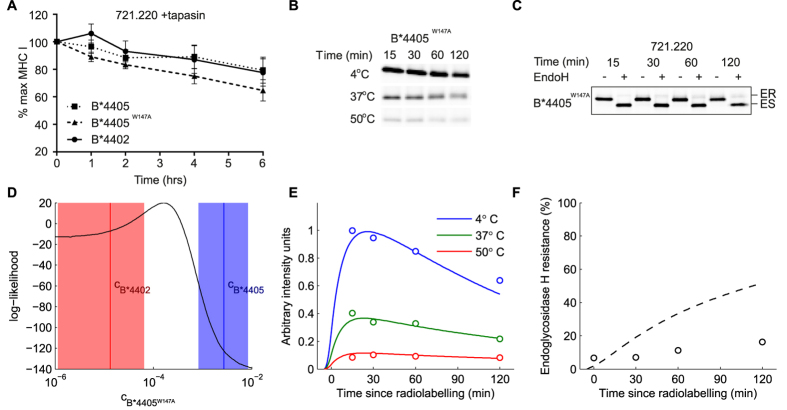
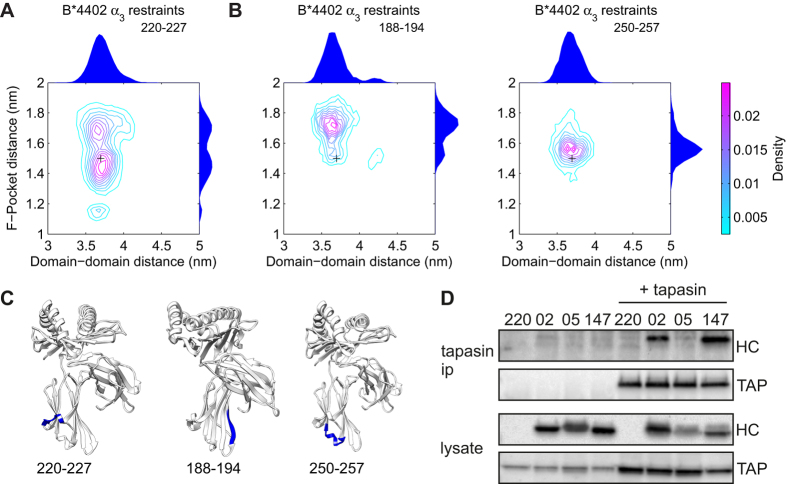
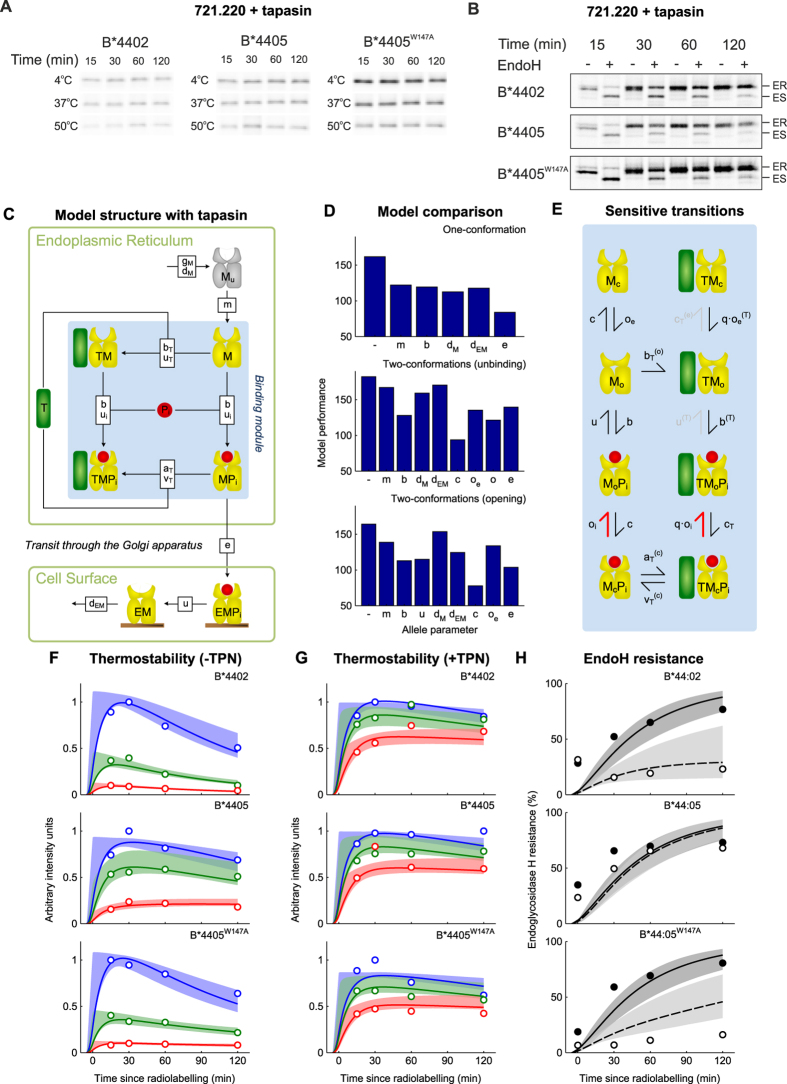
The selection of peptides for presentation at the surface of most nucleated cells by major histocompatibility complex class I molecules (MHC I) is crucial to the immune response in vertebrates. However, the mechanisms of the rapid selection of high affinity peptides by MHC I from amongst thousands of mostly low affinity peptides are not well understood. We developed computational systems models encoding distinct mechanistic hypotheses for two molecules, HLA-B*44:02 (B*4402) and HLA-B*44:05 (B*4405), which differ by a single residue yet lie at opposite ends of the spectrum in their intrinsic ability to select high affinity peptides. We used in vivo biochemical data to infer that a conformational intermediate of MHC I is significant for peptide selection. We used molecular dynamics simulations to show that peptide selector function correlates with protein plasticity, and confirmed this experimentally by altering the plasticity of MHC I with a single point mutation, which altered in vivo selector function in a predictable way. Finally, we investigated the mechanisms by which the co-factor tapasin influences MHC I plasticity. We propose that tapasin modulates MHC I plasticity by dynamically coupling the peptide binding region and α3 domain of MHC I allosterically, resulting in enhanced peptide selector function.
Figures
Similar articles
-
Plasticity of empty major histocompatibility complex class I molecules determines peptide-selector function.Mol Immunol. 2015 Dec;68(2 Pt A):98-101. doi: 10.1016/j.molimm.2015.03.010. Epub 2015 Mar 26. Mol Immunol. 2015. PMID: 25818313 Free PMC article. Review.
-
Molecular architecture of the MHC I peptide-loading complex: one tapasin molecule is essential and sufficient for antigen processing.FASEB J. 2012 Dec;26(12):5071-80. doi: 10.1096/fj.12-217489. Epub 2012 Aug 24. FASEB J. 2012. PMID: 22923333
-
Mechanistic Basis for Epitope Proofreading in the Peptide-Loading Complex.J Immunol. 2015 Nov 1;195(9):4503-13. doi: 10.4049/jimmunol.1501515. Epub 2015 Sep 28. J Immunol. 2015. PMID: 26416272
-
Two polymorphisms facilitate differences in plasticity between two chicken major histocompatibility complex class I proteins.PLoS One. 2014 Feb 20;9(2):e89657. doi: 10.1371/journal.pone.0089657. eCollection 2014. PLoS One. 2014. PMID: 24586943 Free PMC article.
-
Structural aspects of chaperone-mediated peptide loading in the MHC-I antigen presentation pathway.Crit Rev Biochem Mol Biol. 2019 Apr;54(2):164-173. doi: 10.1080/10409238.2019.1610352. Epub 2019 May 14. Crit Rev Biochem Mol Biol. 2019. PMID: 31084439 Free PMC article. Review.
Cited by
-
HLA-B57 micropolymorphism defines the sequence and conformational breadth of the immunopeptidome.Nat Commun. 2018 Nov 8;9(1):4693. doi: 10.1038/s41467-018-07109-w. Nat Commun. 2018. PMID: 30410026 Free PMC article.
-
Molecular mechanism of peptide editing in the tapasin-MHC I complex.Sci Rep. 2016 Jan 12;6:19085. doi: 10.1038/srep19085. Sci Rep. 2016. PMID: 26754481 Free PMC article.
-
Dynamics of MHC-I molecules in the antigen processing and presentation pathway.Curr Opin Immunol. 2021 Jun;70:122-128. doi: 10.1016/j.coi.2021.04.012. Epub 2021 Jun 18. Curr Opin Immunol. 2021. PMID: 34153556 Free PMC article. Review.
-
Dynamic allostery controls the peptide sensitivity of the Ly49C natural killer receptor.J Biol Chem. 2021 Jan-Jun;296:100686. doi: 10.1016/j.jbc.2021.100686. Epub 2021 Apr 21. J Biol Chem. 2021. PMID: 33891944 Free PMC article.
-
A specific amino acid motif of HLA-DRB1 mediates risk and interacts with smoking history in Parkinson's disease.Proc Natl Acad Sci U S A. 2019 Apr 9;116(15):7419-7424. doi: 10.1073/pnas.1821778116. Epub 2019 Mar 25. Proc Natl Acad Sci U S A. 2019. PMID: 30910980 Free PMC article. Clinical Trial.
References
-
- Bjorkman P. J. et al. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature 329, 512–518 (1987). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
