

Molecular signatures of plastic phenotypes in two eusocial insect species with simple societies
- PMID: 26483466
- PMCID: PMC4653166
- DOI: 10.1073/pnas.1515937112
Molecular signatures of plastic phenotypes in two eusocial insect species with simple societies
Abstract
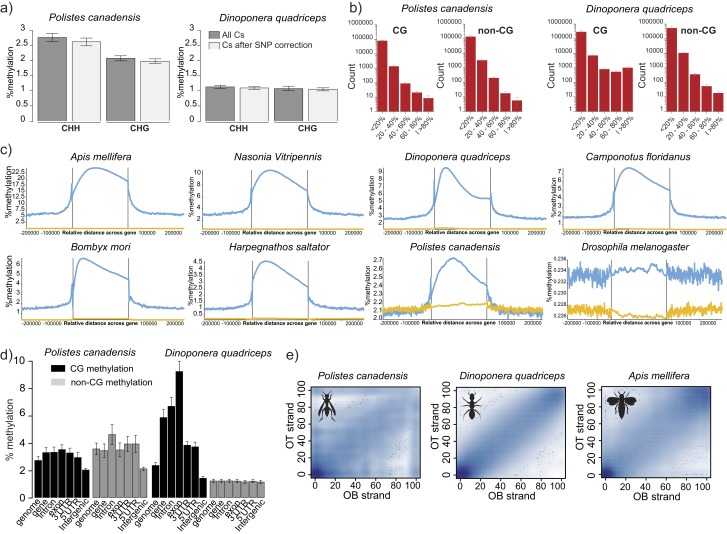
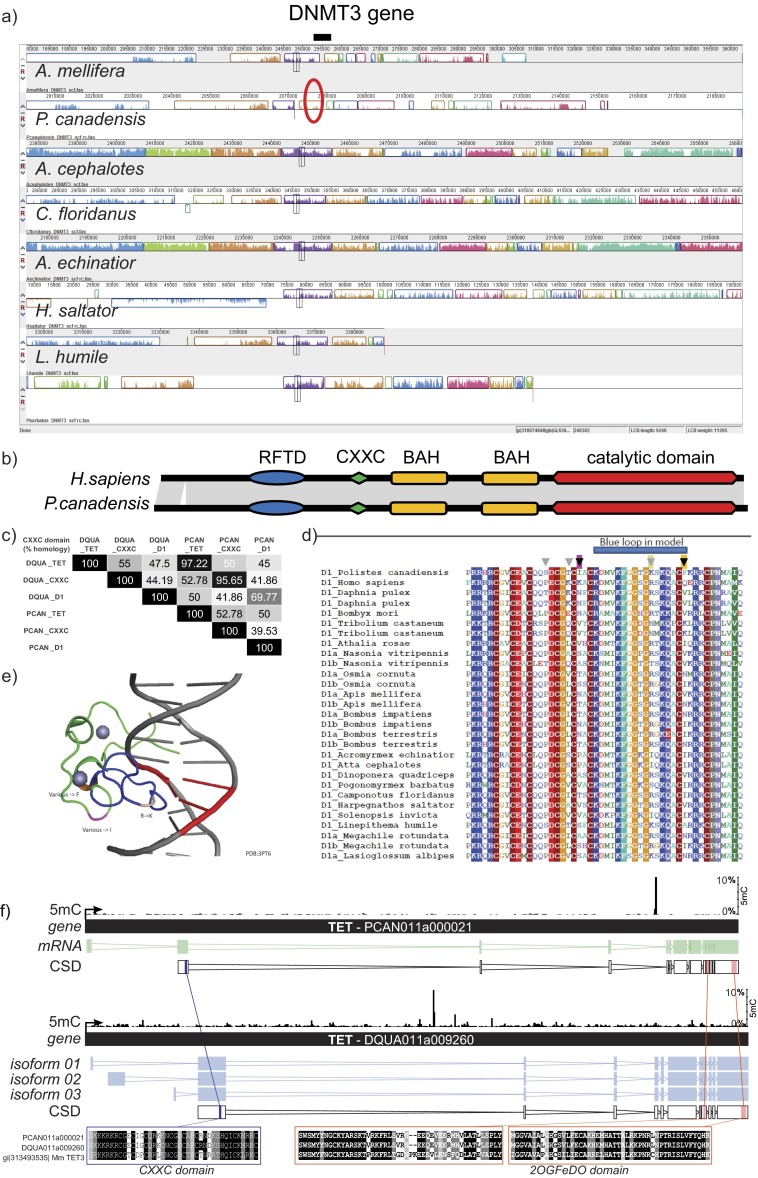
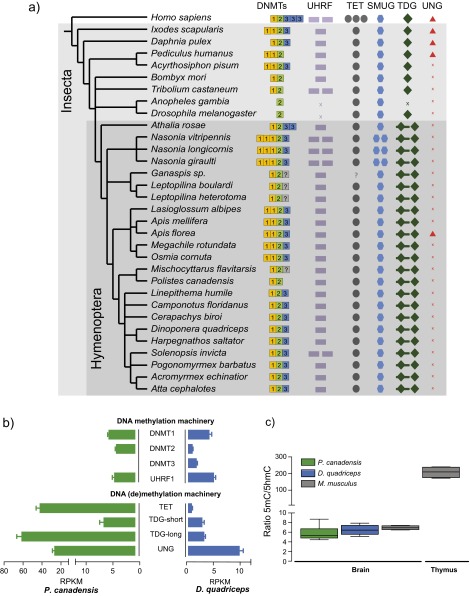
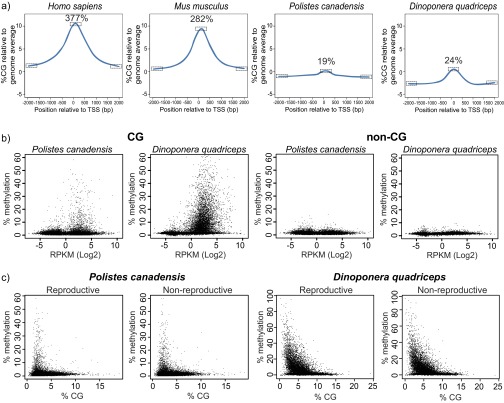
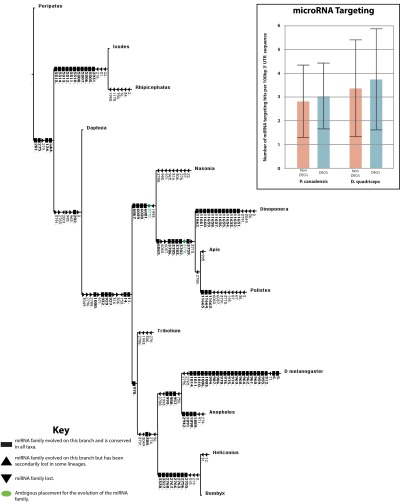
Phenotypic plasticity is important in adaptation and shapes the evolution of organisms. However, we understand little about what aspects of the genome are important in facilitating plasticity. Eusocial insect societies produce plastic phenotypes from the same genome, as reproductives (queens) and nonreproductives (workers). The greatest plasticity is found in the simple eusocial insect societies in which individuals retain the ability to switch between reproductive and nonreproductive phenotypes as adults. We lack comprehensive data on the molecular basis of plastic phenotypes. Here, we sequenced genomes, microRNAs (miRNAs), and multiple transcriptomes and methylomes from individual brains in a wasp (Polistes canadensis) and an ant (Dinoponera quadriceps) that live in simple eusocial societies. In both species, we found few differences between phenotypes at the transcriptional level, with little functional specialization, and no evidence that phenotype-specific gene expression is driven by DNA methylation or miRNAs. Instead, phenotypic differentiation was defined more subtly by nonrandom transcriptional network organization, with roles in these networks for both conserved and taxon-restricted genes. The general lack of highly methylated regions or methylome patterning in both species may be an important mechanism for achieving plasticity among phenotypes during adulthood. These findings define previously unidentified hypotheses on the genomic processes that facilitate plasticity and suggest that the molecular hallmarks of social behavior are likely to differ with the level of social complexity.
Keywords: DNA methylation; genome sequencing; phenotypic plasticity; social evolution; transcriptomes.
Conflict of interest statement
Conflict of interest statement: S.B. is a founder and shareholder of Cambridge Epigenetix Limited, and W.R. is a consultant and shareholder of Cambridge Epigenetix Limited.
Figures

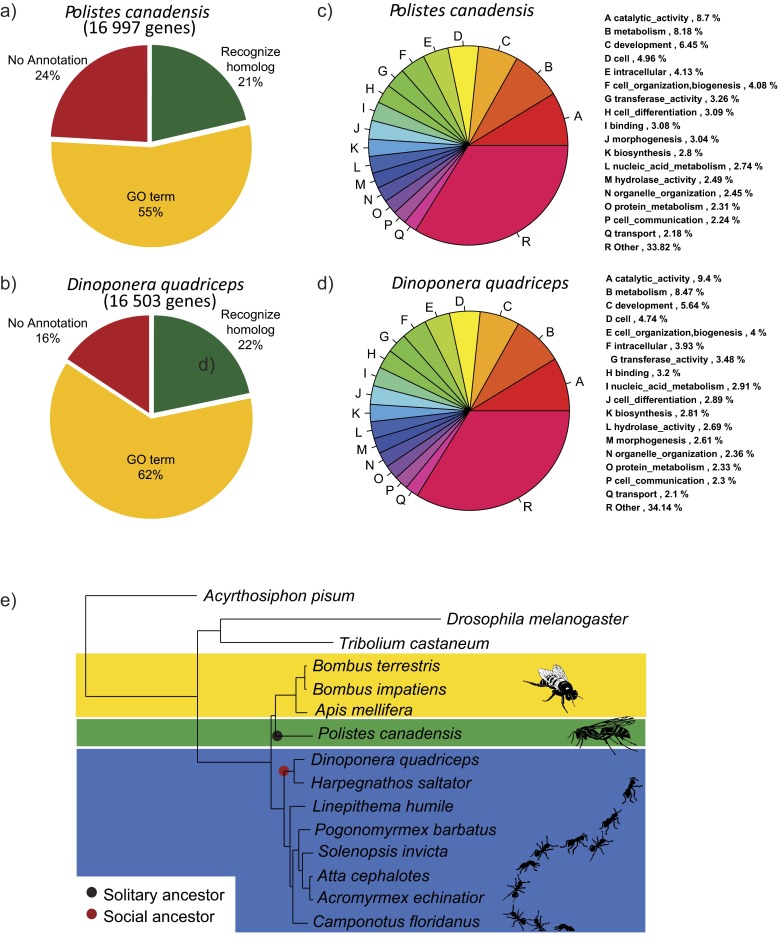
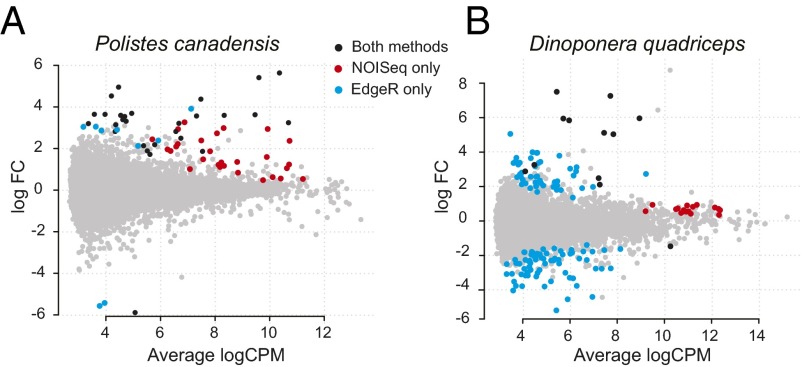
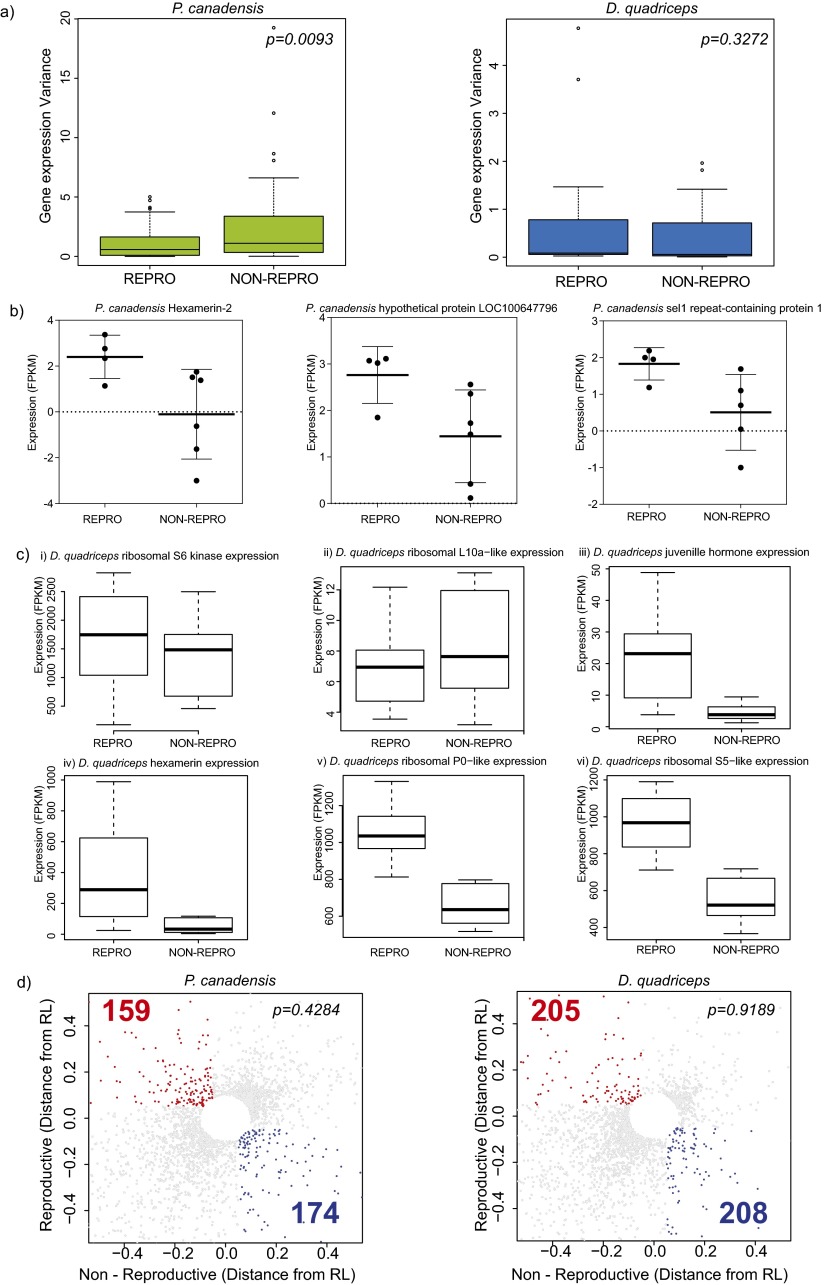
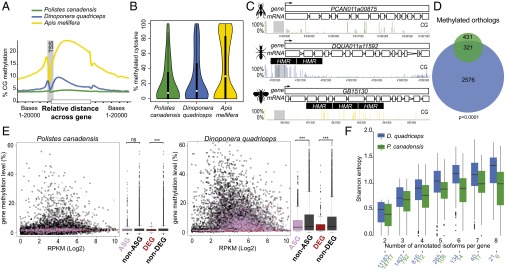


Comment in
-
Genetic architecture of key social trait differs significantly between primitive and advanced eusocial species.Proc Natl Acad Sci U S A. 2015 Nov 10;112(45):13755-6. doi: 10.1073/pnas.1519065112. Epub 2015 Oct 30. Proc Natl Acad Sci U S A. 2015. PMID: 26518508 Free PMC article. No abstract available.
References
-
- West-Eberhard MJ. Developmental Plasticity and Evolution. Oxford Univ Press; New York: 2003.
-
- Pfennig DW, et al. Phenotypic plasticity’s impacts on diversification and speciation. Trends Ecol Evol. 2010;25(8):459–467. - PubMed
-
- Pigliucci M, Murren CJ, Schlichting CD. Phenotypic plasticity and evolution by genetic assimilation. J Exp Biol. 2006;209(Pt 12):2362–2367. - PubMed
-
- Schlichting CD, Wund MA. Phenotypic plasticity and epigenetic marking: An assessment of evidence for genetic accommodation. Evolution. 2014;68(3):656–672. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
