

Reperfusion injury and reactive oxygen species: The evolution of a concept
- PMID: 26484802
- PMCID: PMC4625011
- DOI: 10.1016/j.redox.2015.08.020
Reperfusion injury and reactive oxygen species: The evolution of a concept
Abstract
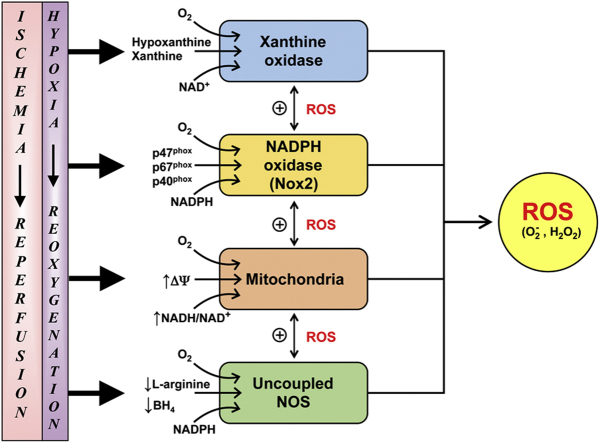
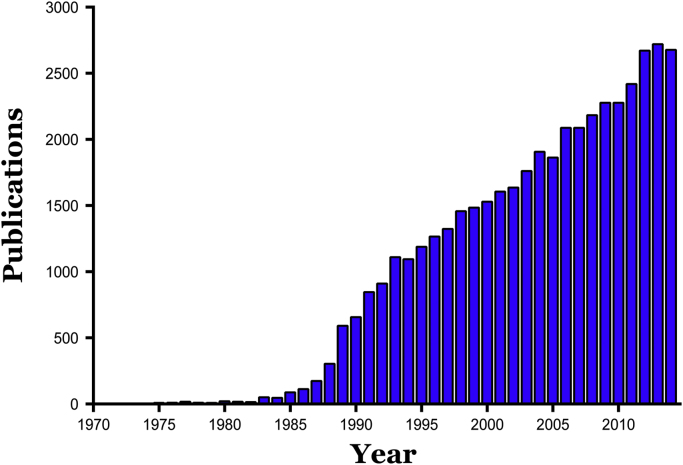
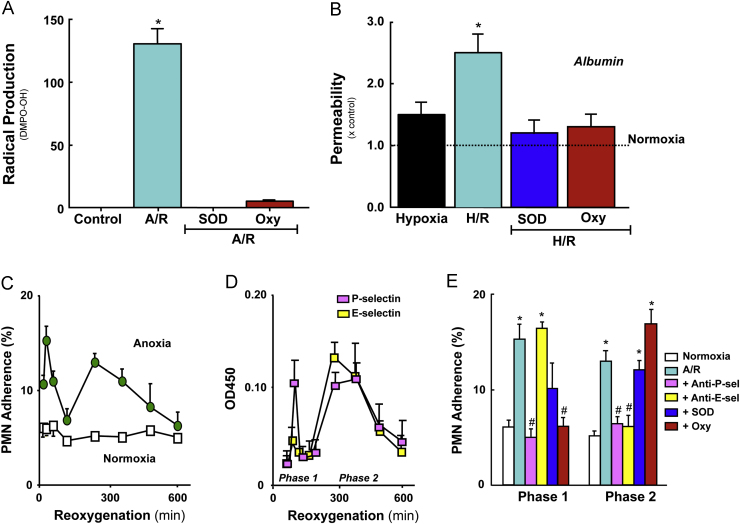
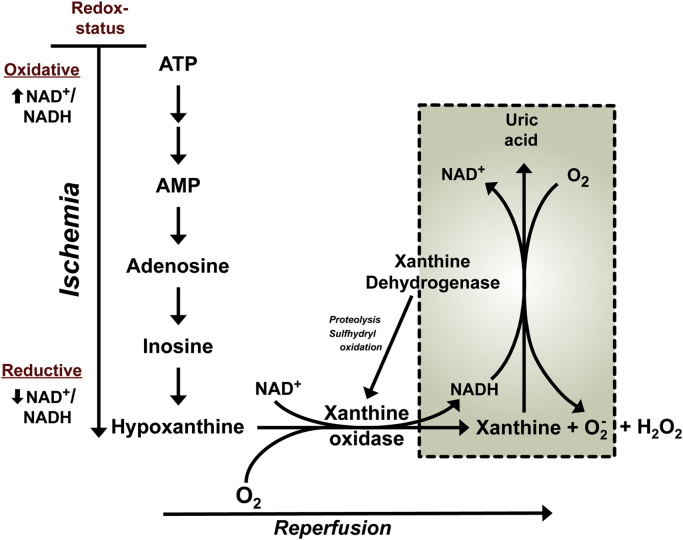
Reperfusion injury, the paradoxical tissue response that is manifested by blood flow-deprived and oxygen-starved organs following the restoration of blood flow and tissue oxygenation, has been a focus of basic and clinical research for over 4-decades. While a variety of molecular mechanisms have been proposed to explain this phenomenon, excess production of reactive oxygen species (ROS) continues to receive much attention as a critical factor in the genesis of reperfusion injury. As a consequence, considerable effort has been devoted to identifying the dominant cellular and enzymatic sources of excess ROS production following ischemia-reperfusion (I/R). Of the potential ROS sources described to date, xanthine oxidase, NADPH oxidase (Nox), mitochondria, and uncoupled nitric oxide synthase have gained a status as the most likely contributors to reperfusion-induced oxidative stress and represent priority targets for therapeutic intervention against reperfusion-induced organ dysfunction and tissue damage. Although all four enzymatic sources are present in most tissues and are likely to play some role in reperfusion injury, priority and emphasis has been given to specific ROS sources that are enriched in certain tissues, such as xanthine oxidase in the gastrointestinal tract and mitochondria in the metabolically active heart and brain. The possibility that multiple ROS sources contribute to reperfusion injury in most tissues is supported by evidence demonstrating that redox-signaling enables ROS produced by one enzymatic source (e.g., Nox) to activate and enhance ROS production by a second source (e.g., mitochondria). This review provides a synopsis of the evidence implicating ROS in reperfusion injury, the clinical implications of this phenomenon, and summarizes current understanding of the four most frequently invoked enzymatic sources of ROS production in post-ischemic tissue.
Keywords: Hypoxia-reoxygenation; Ischemia-reperfusion; Mitochondria; NADPH oxidase; Uncoupled nitric oxide synthase; Xanthine oxidase.
Copyright © 2015 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
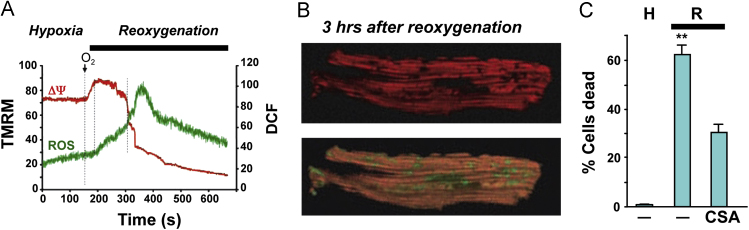

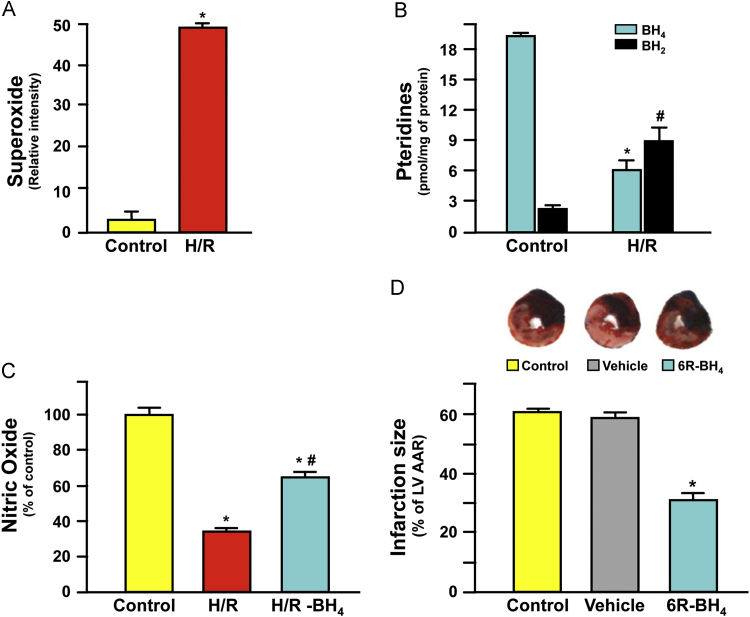
References
-
- Carden D.L., Granger D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000;190:255–266. - PubMed
-
- Raedschelders K., Ansley D.M., Chen D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012;133:230–255. - PubMed
-
- Costantino C., Corday E., Lang T.W., Meerbaum S., Brasch J., Kaplan L., Rubins S., Gold H., Osher J. Revascularization after 3 h of coronary arterial occlusion: effects on regional cardiac metabolic function and infarct size. Am. J. Cardiol. 1975;36:368–384. - PubMed
-
- Reimer K.A., Lowe J.E., Rasmussen M.M., Jennings R.B. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation. 1977;56:786–794. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
