

The human skin double-stranded DNA virome: topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome
- PMID: 26489866
- PMCID: PMC4620475
- DOI: 10.1128/mBio.01578-15
The human skin double-stranded DNA virome: topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome
Abstract
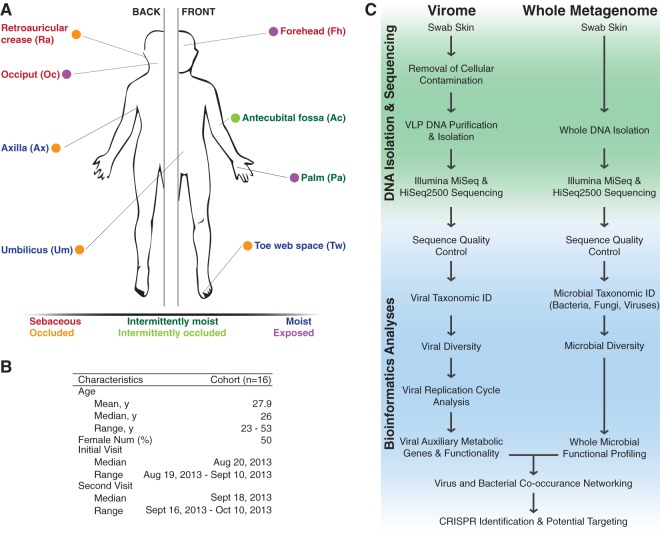
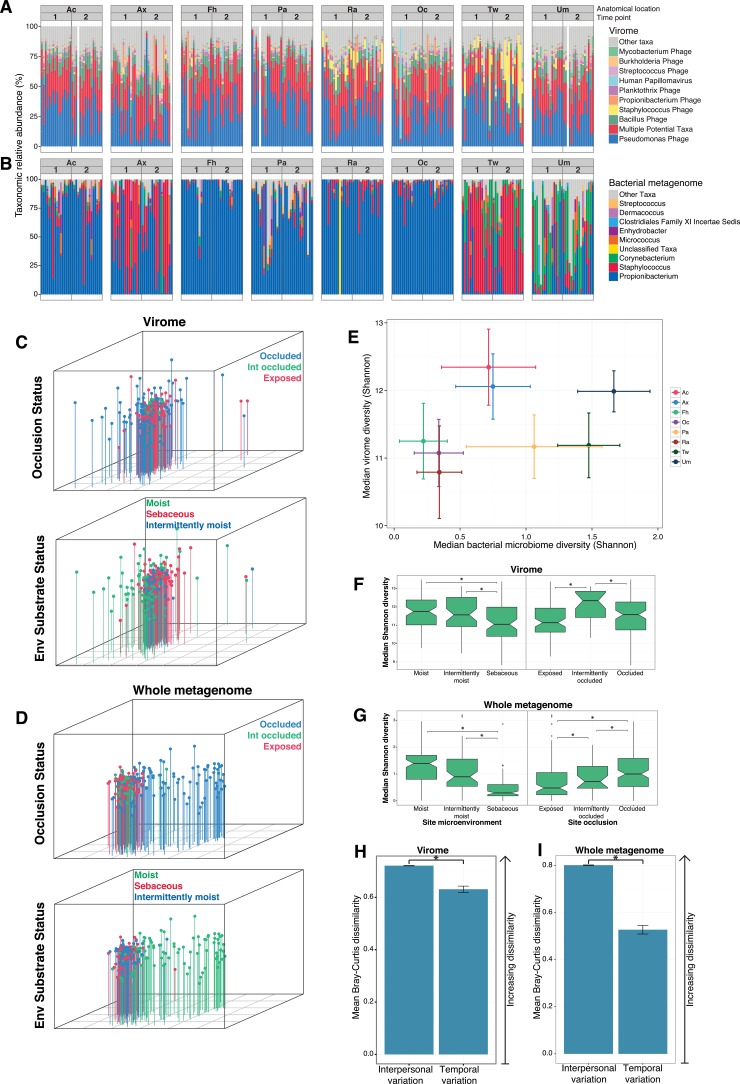
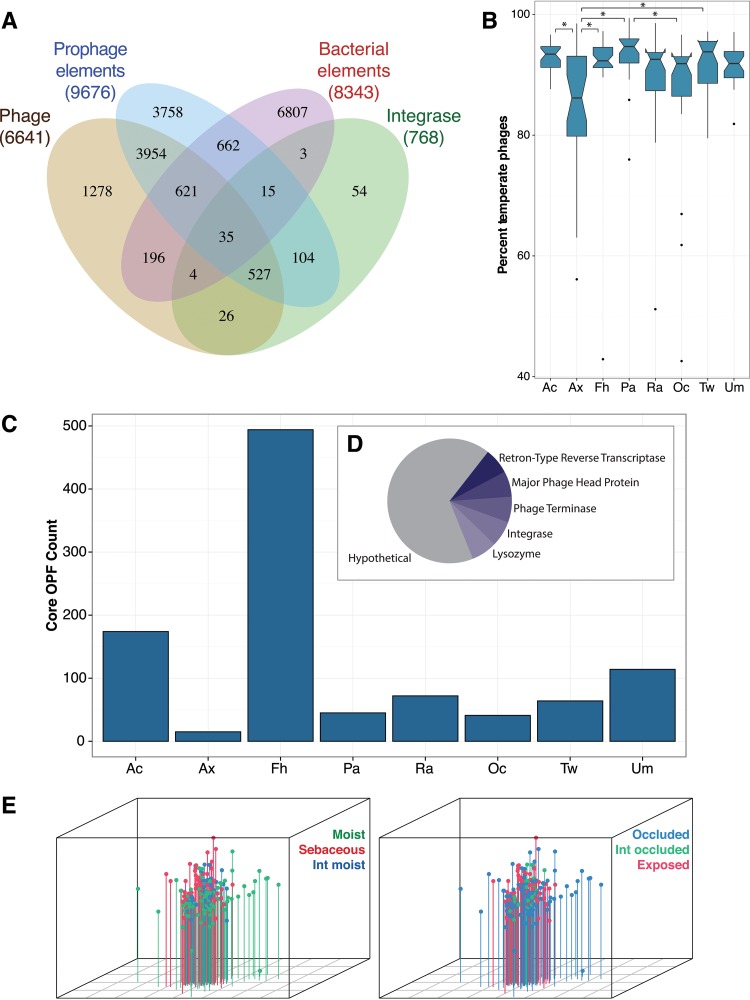
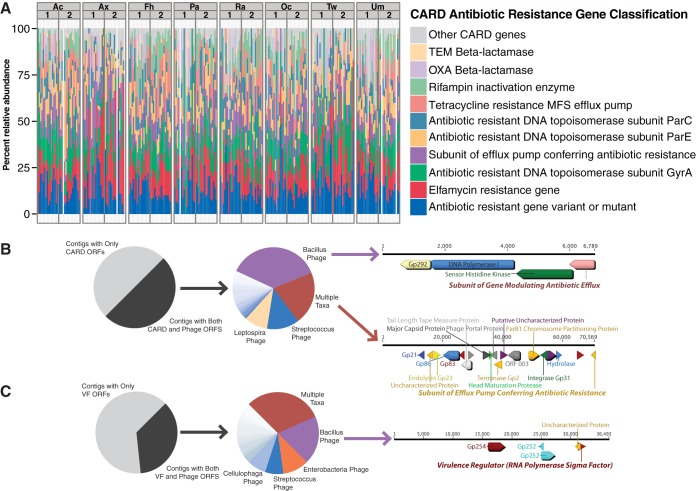
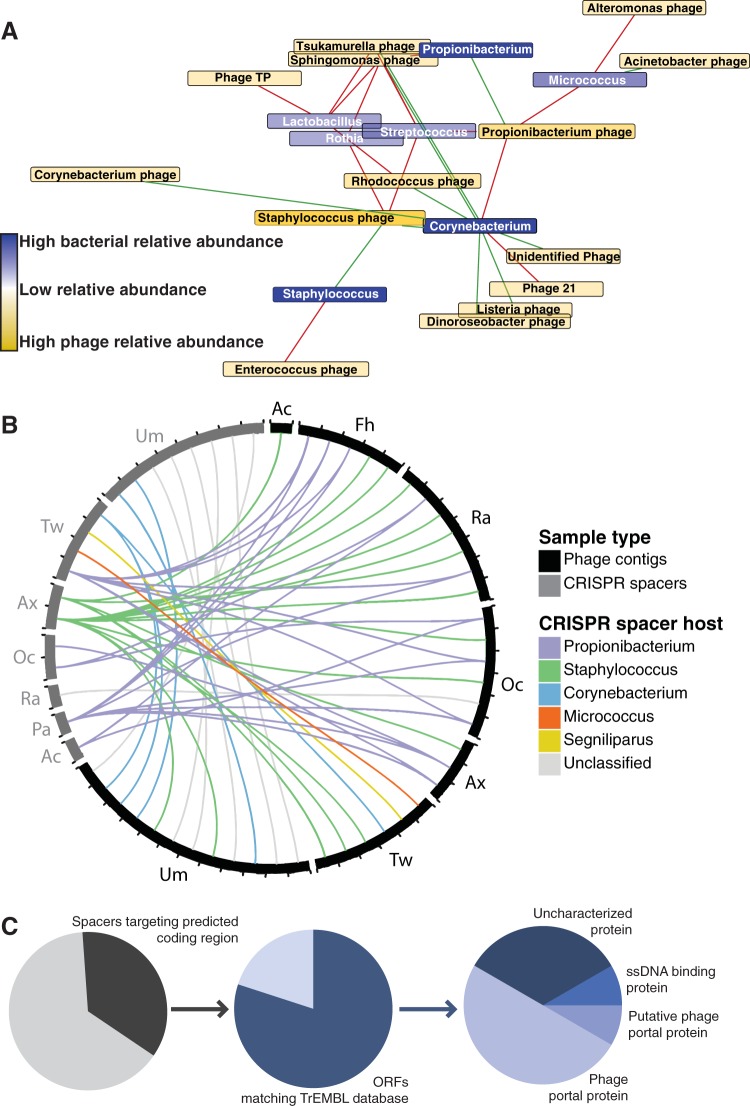
Viruses make up a major component of the human microbiota but are poorly understood in the skin, our primary barrier to the external environment. Viral communities have the potential to modulate states of cutaneous health and disease. Bacteriophages are known to influence the structure and function of microbial communities through predation and genetic exchange. Human viruses are associated with skin cancers and a multitude of cutaneous manifestations. Despite these important roles, little is known regarding the human skin virome and its interactions with the host microbiome. Here we evaluated the human cutaneous double-stranded DNA virome by metagenomic sequencing of DNA from purified virus-like particles (VLPs). In parallel, we employed metagenomic sequencing of the total skin microbiome to assess covariation and infer interactions with the virome. Samples were collected from 16 subjects at eight body sites over 1 month. In addition to the microenviroment, which is known to partition the bacterial and fungal microbiota, natural skin occlusion was strongly associated with skin virome community composition. Viral contigs were enriched for genes indicative of a temperate phage replication style and also maintained genes encoding potential antibiotic resistance and virulence factors. CRISPR spacers identified in the bacterial DNA sequences provided a record of phage predation and suggest a mechanism to explain spatial partitioning of skin phage communities. Finally, we modeled the structure of bacterial and phage communities together to reveal a complex microbial environment with a Corynebacterium hub. These results reveal the previously underappreciated diversity, encoded functions, and viral-microbial dynamic unique to the human skin virome.
Importance: To date, most cutaneous microbiome studies have focused on bacterial and fungal communities. Skin viral communities and their relationships with their hosts remain poorly understood despite their potential to modulate states of cutaneous health and disease. Previous studies employing whole-metagenome sequencing without purification for virus-like particles (VLPs) have provided some insight into the viral component of the skin microbiome but have not completely characterized these communities or analyzed interactions with the host microbiome. Here we present an optimized virus purification technique and corresponding analysis tools for gaining novel insights into the skin virome, including viral "dark matter," and its potential interactions with the host microbiome. The work presented here establishes a baseline of the healthy human skin virome and is a necessary foundation for future studies examining viral perturbations in skin health and disease.
Copyright © 2015 Hannigan et al.
Figures
References
-
- Rodriguez-Brito B, Li L, Wegley L, Furlan M, Angly F, Breitbart M, Buchanan J, Desnues C, Dinsdale E, Edwards R, Felts B, Haynes M, Liu H, Lipson D, Mahaffy J, Martin-Cuadrado AB, Mira A, Nulton J, Pašić L, Rayhawk S, Rodriguez-Mueller J, Rodriguez-Valera F, Salamon P, Srinagesh S, Thingstad TF, Tran T, Thurber RV, Willner D, Youle M, Rohwer F. 2010. Viral and microbial community dynamics in four aquatic environments. ISME J 4:739–751. doi: 10.1038/ismej.2010.1. - DOI - PubMed
-
- Tyson GW, Banfield JF. 2008. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ Microbiol 10:200–207. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
