

Complement activation, regulation, and molecular basis for complement-related diseases
- PMID: 26489954
- PMCID: PMC4682646
- DOI: 10.15252/embj.201591881
Complement activation, regulation, and molecular basis for complement-related diseases
Abstract
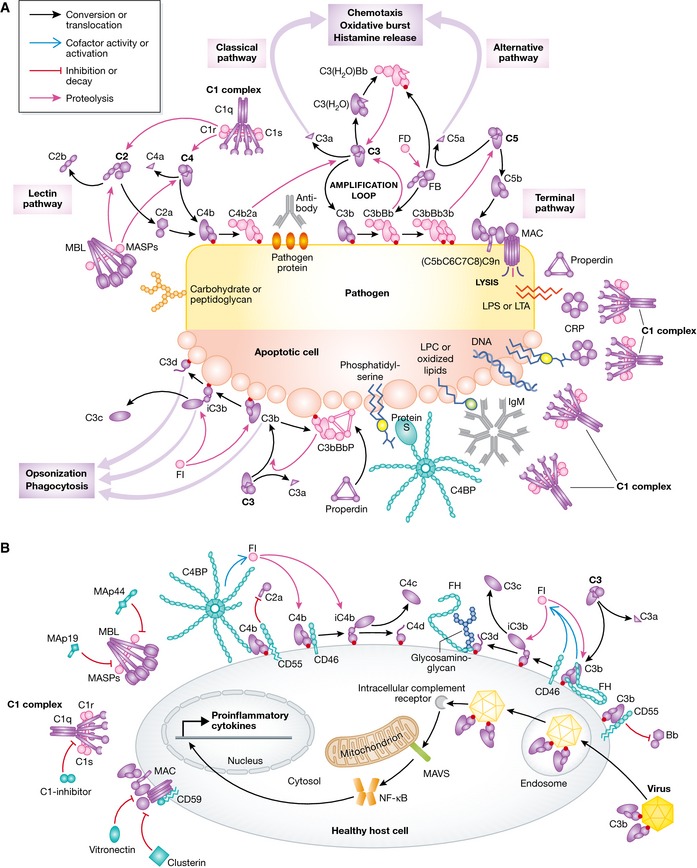
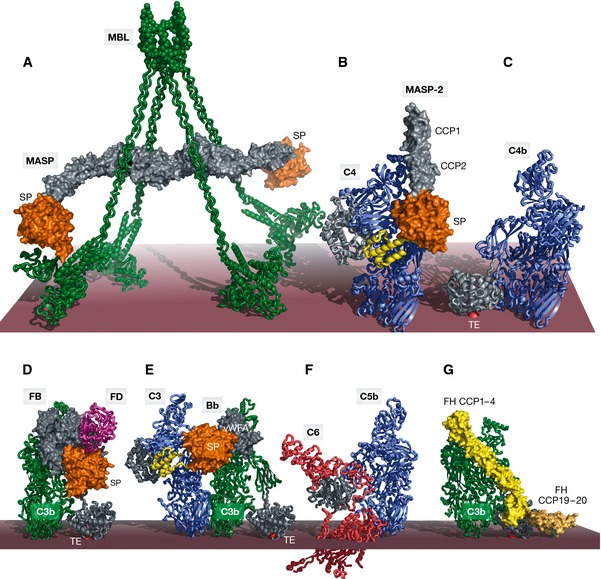
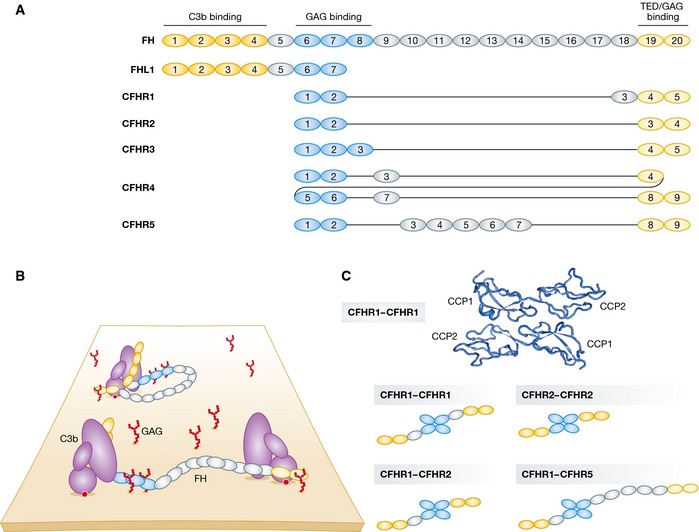
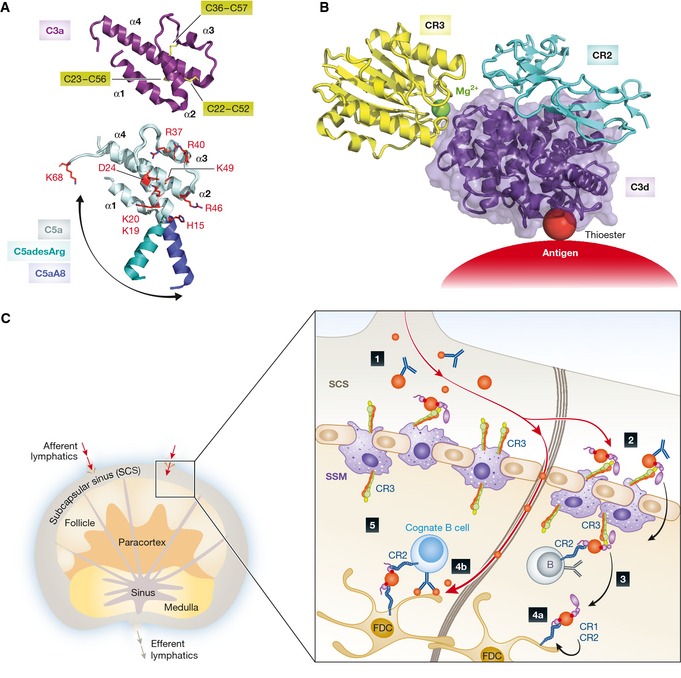
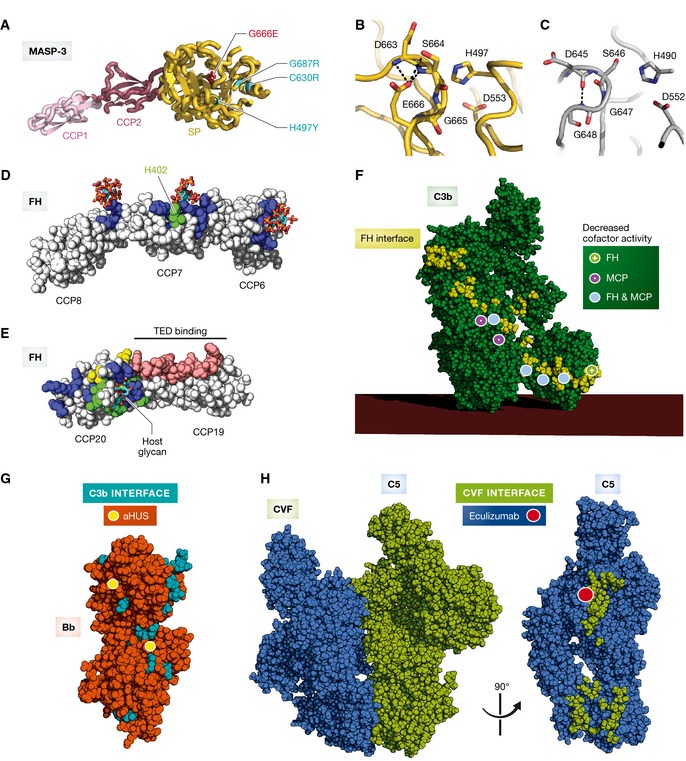
The complement system is an essential element of the innate immune response that becomes activated upon recognition of molecular patterns associated with microorganisms, abnormal host cells, and modified molecules in the extracellular environment. The resulting proteolytic cascade tags the complement activator for elimination and elicits a pro-inflammatory response leading to recruitment and activation of immune cells from both the innate and adaptive branches of the immune system. Through these activities, complement functions in the first line of defense against pathogens but also contributes significantly to the maintenance of homeostasis and prevention of autoimmunity. Activation of complement and the subsequent biological responses occur primarily in the extracellular environment. However, recent studies have demonstrated autocrine signaling by complement activation in intracellular vesicles, while the presence of a cytoplasmic receptor serves to detect complement-opsonized intracellular pathogens. Furthermore, breakthroughs in both functional and structural studies now make it possible to describe many of the intricate molecular mechanisms underlying complement activation and the subsequent downstream events, as well as its cross talk with, for example, signaling pathways, the coagulation system, and adaptive immunity. We present an integrated and updated view of complement based on structural and functional data and describe the new roles attributed to complement. Finally, we discuss how the structural and mechanistic understanding of the complement system rationalizes the genetic defects conferring uncontrolled activation or other undesirable effects of complement.
Keywords: complement; inflammation; innate immunity; proteolytic regulation; structural biology.
© 2015 The Authors.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
