

Antiproteinuric therapy and Fabry nephropathy: factors associated with preserved kidney function during agalsidase-beta therapy
- PMID: 26490103
- PMCID: PMC4717450
- DOI: 10.1136/jmedgenet-2015-103471
Antiproteinuric therapy and Fabry nephropathy: factors associated with preserved kidney function during agalsidase-beta therapy
Abstract
Background: Nephropathy is an important feature of classical Fabry disease, which results in alpha-galactosidase A deficiency and cellular globotriaosylceramide accumulation. We report the safety and efficacy of antiproteinuric therapy with ACE inhibitors or angiotensin II receptor blockers (ARBs) in a study of classical Fabry patients receiving recombinant agalsidase-beta therapy.
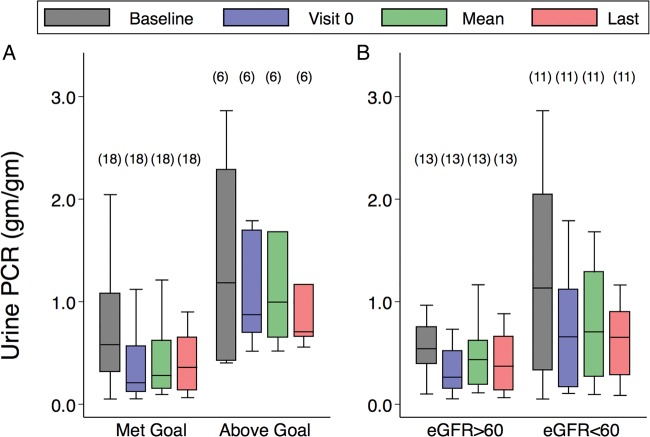
Methods and design: The goal was maintenance of urine protein to creatinine ratio (UPCR) <0.5 g/g or a 50% reduction in baseline UPCR for 24 patients at eight study sites. The change in estimated glomerular filtration rate (eGFR) was assessed over 21 months of treatment.
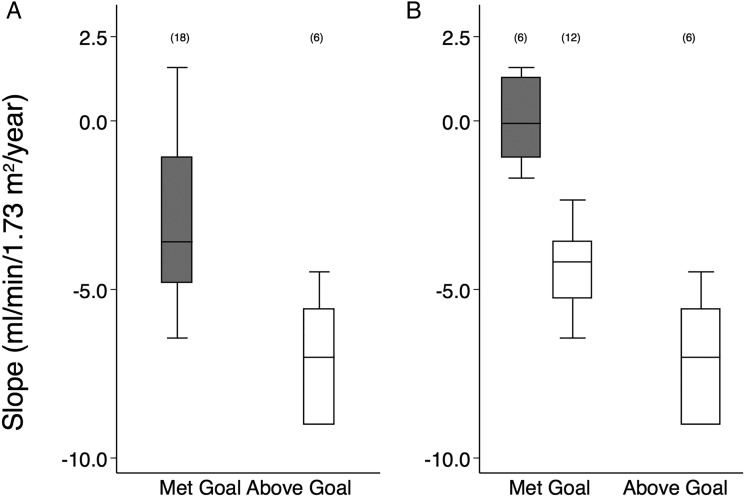
Results: 18 out of 24 patients achieved the UPCR goal with eGFR slopes that were significantly better than six patients who did not achieve the UPCR goal (-3.6 (-4.8 to -1.1) versus -7.0 (-9.0 to -5.6) mL/min/1.73 m(2)/year, respectively, p=0.018). Despite achieving the UPCR goal, 67% (12/18 patients) still progressed with an eGFR slope <-2 mL/min/1.73 m(2)/year. Regression analysis showed that increased age at initiation of agalsidase-beta therapy was significantly associated with worsened kidney outcome. Hypotension and hyperkalaemia occurred in seven and eight patients, respectively, which required modification of antiproteinuric therapy but was not associated with serious adverse events.
Conclusions: This study documents the effectiveness of agalsidase-beta (1 mg/kg/2 weeks) and antiproteinuric therapy with ACE inhibitors and/or ARB in patients with severe Fabry nephropathy. Patients had preservation of kidney function if agalsidase-beta treatment was initiated at a younger age, and UPCR maintained at or below 0.5 g/g with antiproteinuric therapy.
Trial registration number: NCT00446862.
Keywords: Clinical genetics; Metabolic disorders; Renal Medicine.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/
Figures
References
-
- Desnick RJ, Ioannou YA, Eng CM. Alpha-galactosidase A deficiency: fabry disease. In: Scriver C, Beaudet A, Sly W, et al., eds The metabolic bases of inherited disease. 8th edn New York: McGraw-Hill, 2001:3733–74.
-
- Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, Sims K, Waldek S, Pastores GM, Lee P, Eng CM, Marodi L, Stanford KE, Breunig F, Wanner C, Warnock DG, Lemay RM, Germain DP. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol Genet Metab 2008;93:112–28. 10.1016/j.ymgme.2007.09.013 - DOI - PubMed
-
- Dobrovolny R, Dvorakova L, Ledvinova J, Magage S, Bultas J, Lubanda JC, Elleder M, Karetova D, Pavlikova M, Hrebicek M. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population . J Mol Med 2005;83:647–54. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous