

Induction of Kaposi's Sarcoma-Associated Herpesvirus-Encoded Viral Interleukin-6 by X-Box Binding Protein 1
- PMID: 26491160
- PMCID: PMC4702535
- DOI: 10.1128/JVI.01192-15
Induction of Kaposi's Sarcoma-Associated Herpesvirus-Encoded Viral Interleukin-6 by X-Box Binding Protein 1
Abstract
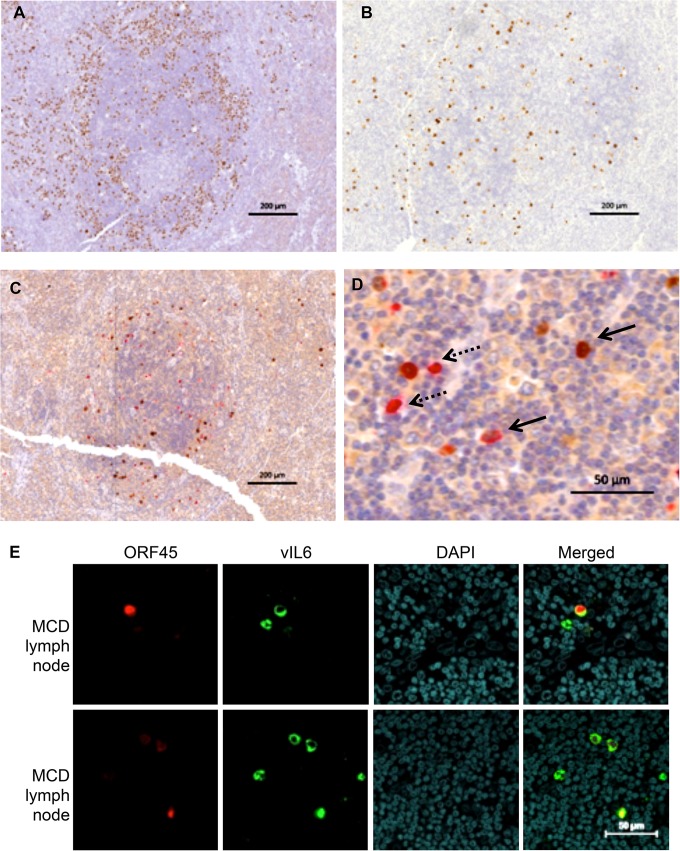
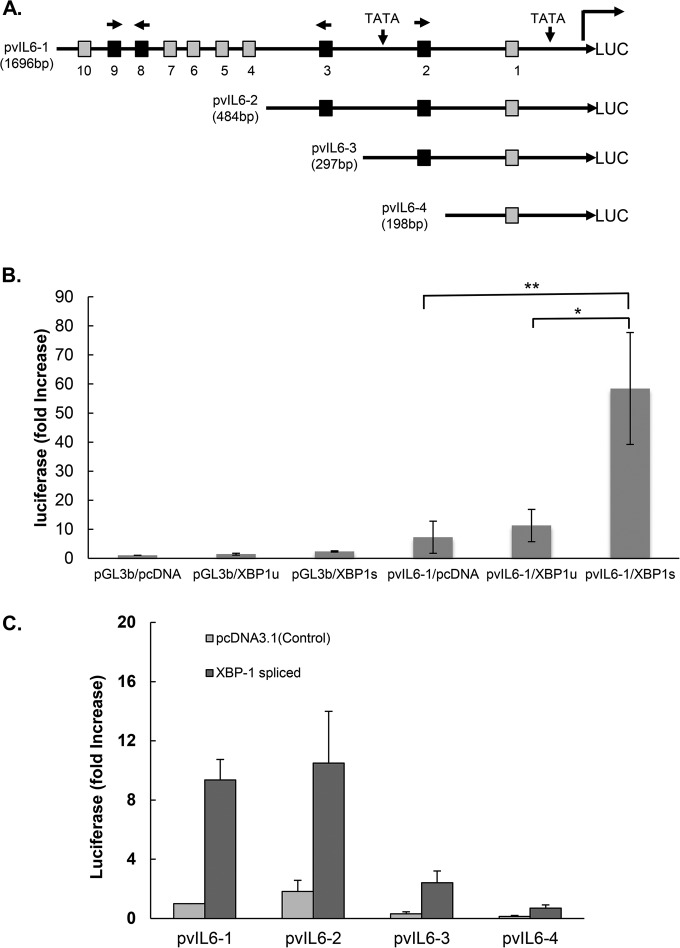
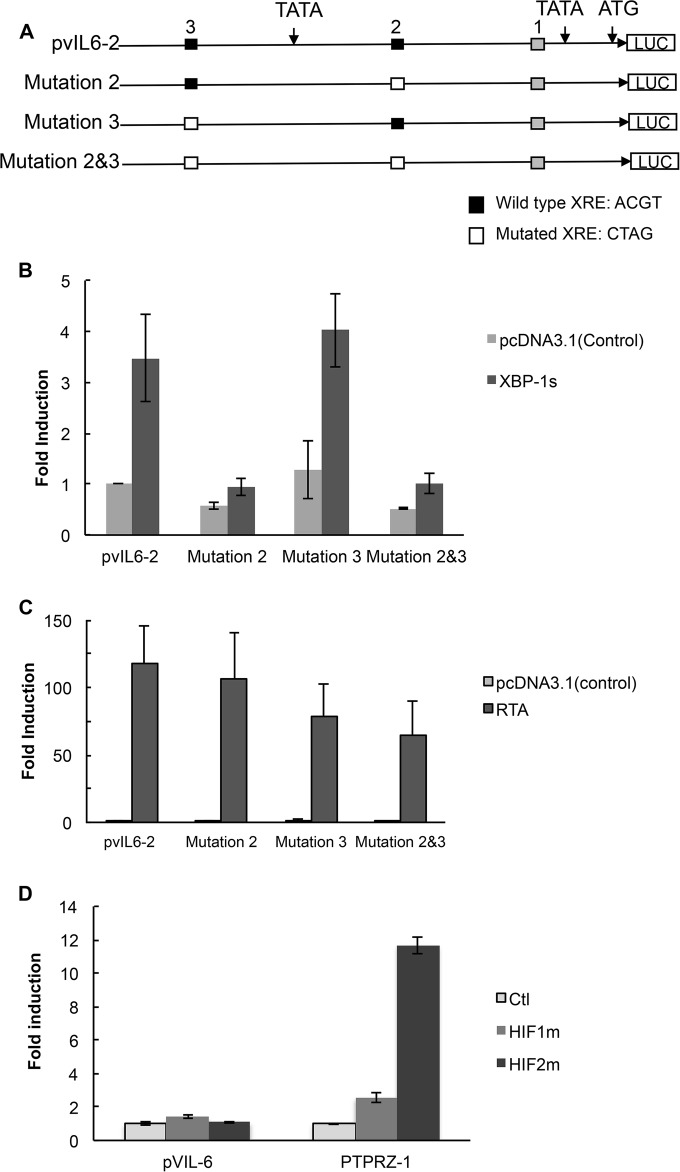
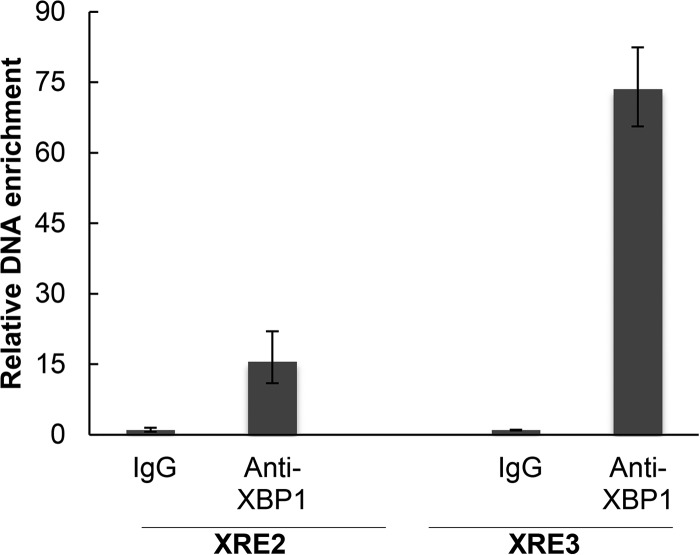
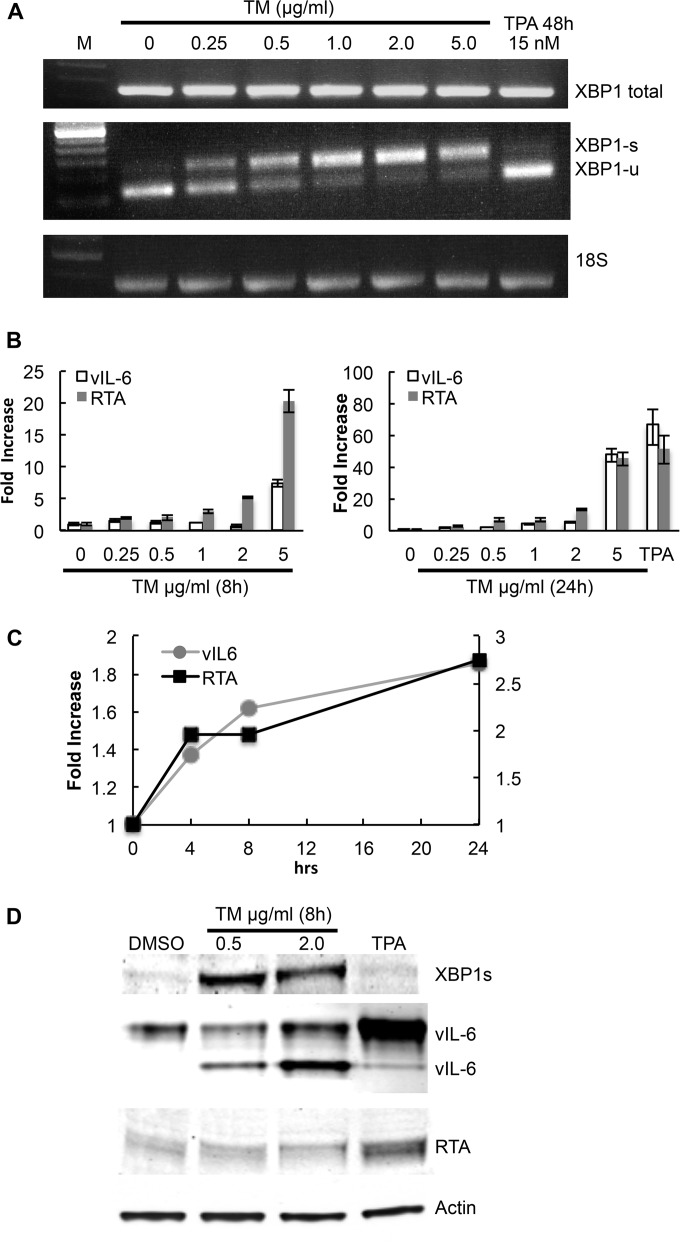
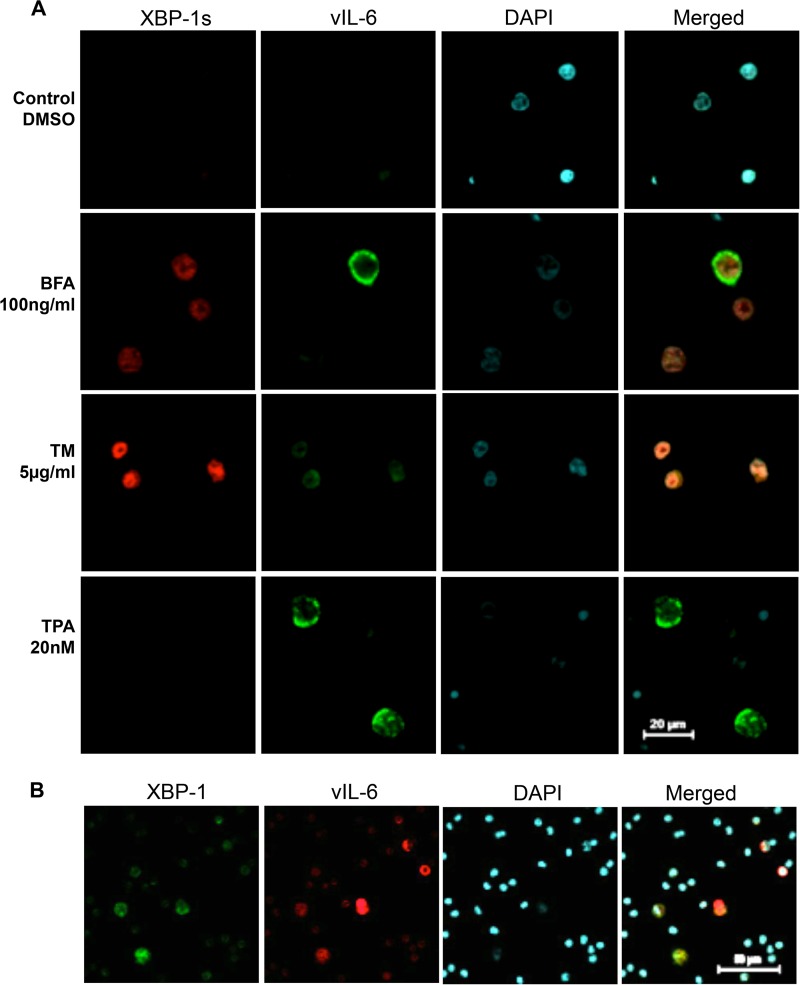
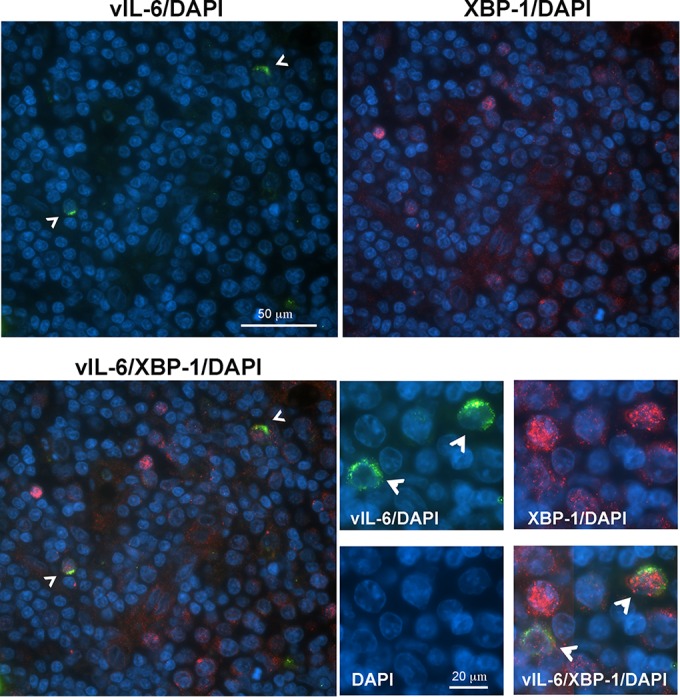
Kaposi's sarcoma-associated herpesvirus (KSHV) is the causative agent for Kaposi sarcoma (KS), primary effusion lymphoma (PEL), and a subset of multicentric Castleman disease (MCD). The KSHV life cycle has two principal gene repertoires, latent and lytic. KSHV viral interleukin-6 (vIL-6), an analog of human IL-6, is usually lytic; production of vIL-6 by involved plasmablasts is a central feature of KSHV-MCD. vIL-6 also plays a role in PEL and KS. We show that a number of plasmablasts from lymph nodes of patients with KSHV-MCD express vIL-6 but not ORF45, a KSHV lytic gene. We further show that vIL-6 is directly induced by the spliced (active) X-box binding protein-1 (XBP-1s), a transcription factor activated by endoplasmic reticulum (ER) stress and differentiation of B cells in lymph nodes. The promoter region of vIL-6 contains several potential XBP-response elements (XREs), and two of these elements in particular mediate the effect of XBP-1s. Mutation of these elements abrogates the response to XBP-1s but not to the KSHV replication and transcription activator (RTA). Also, XBP-1s binds to the vIL-6 promoter in the region of these XREs. Exposure of PEL cells to a chemical inducer of XBP-1s can induce vIL-6. Patient-derived PEL tumor cells that produce vIL-6 frequently coexpress XBP-1, and immunofluorescence staining of involved KSHV-MCD lymph nodes reveals that most plasmablasts expressing vIL-6 also coexpress XBP-1. These results provide evidence that XBP-1s is a direct activator of KSHV vIL-6 and that this is an important step in the pathogenesis of KSHV-MCD and PEL.
Importance: Kaposi sarcoma herpesvirus (KSHV)-associated multicentric Castleman disease (KSHV-MCD) is characterized by severe inflammatory symptoms caused by an excess of cytokines, particularly KSHV-encoded viral interleukin-6 (vIL-6) produced by lymph node plasmablasts. vIL-6 is usually a lytic gene. We show that a number of KSHV-MCD lymph node plasmablasts express vIL-6 but do not have full lytic KSHV replication. Differentiating lymph node B cells express spliced (active) X-box binding protein-1 (XBP-1s). We show that XBP-1s binds to the promoter of vIL-6 and can directly induce production of vIL-6 through X-box protein response elements on the vIL-6 promoter region. We further show that chemical inducers of XBP-1s can upregulate production of vIL-6. Finally, we show that most vIL-6-producing plasmablasts from lymph nodes of KSHV-MCD patients coexpress XBP-1s. These results demonstrate that XBP-1s can directly induce vIL-6 and provide evidence that this is a key step in the pathogenesis of KSHV-MCD and other KSHV-induced diseases.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Induction of Kaposi's Sarcoma-Associated Herpesvirus-Encoded Thymidine Kinase (ORF21) by X-Box Binding Protein 1.J Virol. 2020 Feb 14;94(5):e01555-19. doi: 10.1128/JVI.01555-19. Print 2020 Feb 14. J Virol. 2020. PMID: 31801863 Free PMC article.
-
X box binding protein XBP-1s transactivates the Kaposi's sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency.J Virol. 2007 Dec;81(24):13578-86. doi: 10.1128/JVI.01663-07. Epub 2007 Oct 10. J Virol. 2007. PMID: 17928342 Free PMC article.
-
X-box binding protein 1 induces the expression of the lytic cycle transactivator of Kaposi's sarcoma-associated herpesvirus but not Epstein-Barr virus in co-infected primary effusion lymphoma.J Gen Virol. 2011 Feb;92(Pt 2):421-31. doi: 10.1099/vir.0.025494-0. Epub 2010 Oct 27. J Gen Virol. 2011. PMID: 20980528 Free PMC article.
-
Pathological Features of Kaposi's Sarcoma-Associated Herpesvirus Infection.Adv Exp Med Biol. 2018;1045:357-376. doi: 10.1007/978-981-10-7230-7_16. Adv Exp Med Biol. 2018. PMID: 29896675 Review.
-
KSHV-associated multicentric Castleman disease: A tangle of different entities requiring multitarget treatment strategies.Int J Cancer. 2015 Jul 15;137(2):251-61. doi: 10.1002/ijc.28923. Epub 2014 May 5. Int J Cancer. 2015. PMID: 24771491 Review.
Cited by
-
HIV-associated Kaposi sarcoma and related diseases.AIDS. 2017 Sep 10;31(14):1903-1916. doi: 10.1097/QAD.0000000000001567. AIDS. 2017. PMID: 28609402 Free PMC article. Review.
-
NAT10-dependent N4-acetylcytidine modification mediates PAN RNA stability, KSHV reactivation, and IFI16-related inflammasome activation.Nat Commun. 2023 Oct 10;14(1):6327. doi: 10.1038/s41467-023-42135-3. Nat Commun. 2023. PMID: 37816771 Free PMC article.
-
Multicentric Castleman disease and the evolution of the concept.Pathologica. 2021 Oct;113(5):339-353. doi: 10.32074/1591-951X-351. Pathologica. 2021. PMID: 34837092 Free PMC article. Review.
-
BiP/GRP78 is a pro-viral factor for diverse dsDNA viruses that promotes the survival and proliferation of cells upon KSHV infection.PLoS Pathog. 2024 Oct 29;20(10):e1012660. doi: 10.1371/journal.ppat.1012660. eCollection 2024 Oct. PLoS Pathog. 2024. PMID: 39471213 Free PMC article.
-
HIV-associated cancers and lymphoproliferative disorders caused by Kaposi sarcoma herpesvirus and Epstein-Barr virus.Clin Microbiol Rev. 2024 Sep 12;37(3):e0002223. doi: 10.1128/cmr.00022-23. Epub 2024 Jun 20. Clin Microbiol Rev. 2024. PMID: 38899877 Free PMC article. Review.
References
-
- Dupin N, Diss TL, Kellam P, Tulliez M, Du MQ, Sicard D, Weiss RA, Isaacson PG, Boshoff C. 2000. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 95:1406–1412. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
