

Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses
- PMID: 26491167
- PMCID: PMC4702705
- DOI: 10.1128/JVI.02036-15
Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses
Abstract
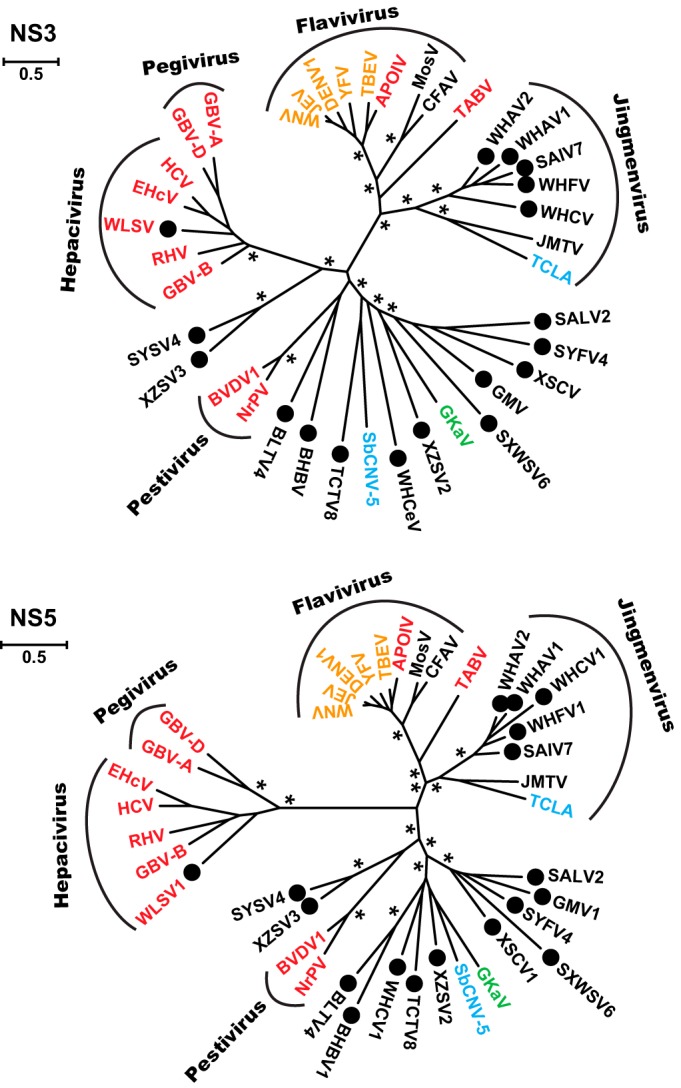
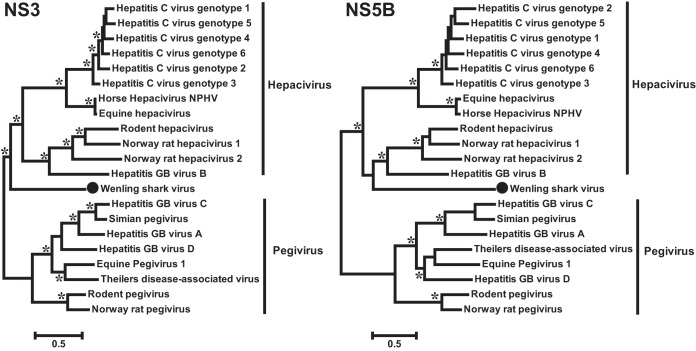
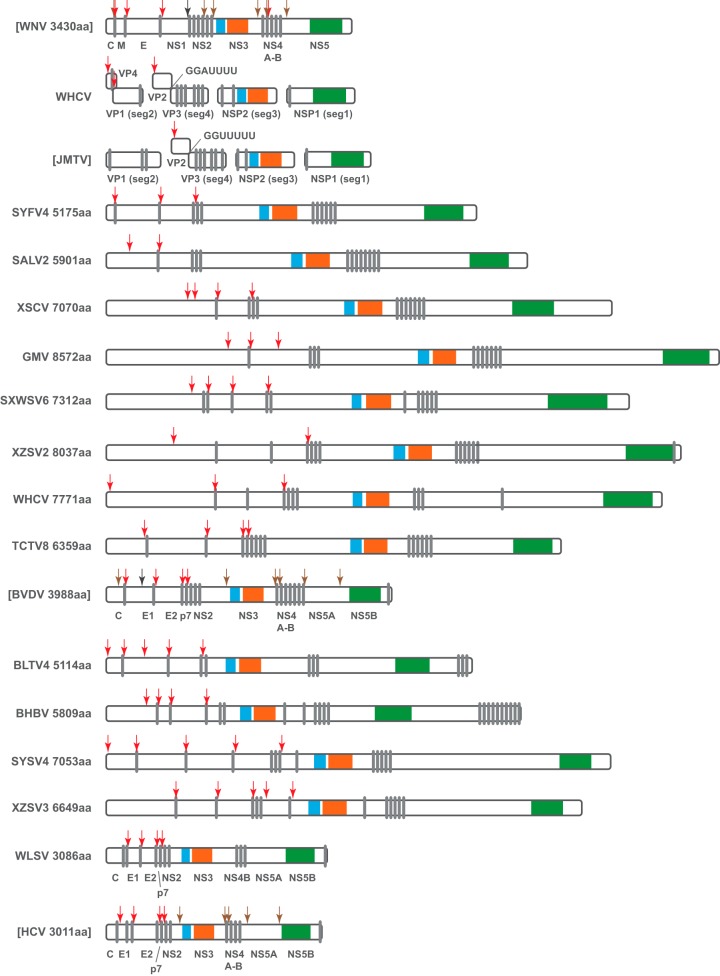
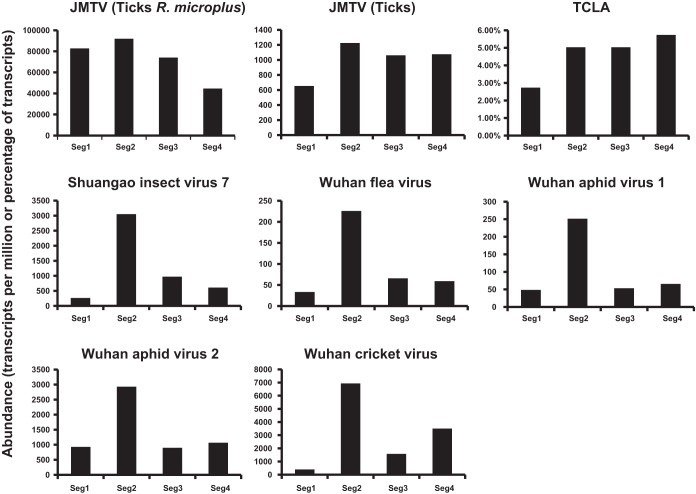
Viruses of the family Flaviviridae are important pathogens of humans and other animals and are currently classified into four genera. To better understand their diversity, evolutionary history, and genomic flexibility, we used transcriptome sequencing (RNA-seq) to search for the viruses related to the Flaviviridae in a range of potential invertebrate and vertebrate hosts. Accordingly, we recovered the full genomes of five segmented jingmenviruses and 12 distant relatives of the known Flaviviridae ("flavi-like" viruses) from a range of arthropod species. Although these viruses are highly divergent, they share a similar genomic plan and common ancestry with the Flaviviridae in the NS3 and NS5 regions. Remarkably, although these viruses fill in major gaps in the phylogenetic diversity of the Flaviviridae, genomic comparisons reveal important changes in genome structure, genome size, and replication/gene regulation strategy during evolutionary history. In addition, the wide diversity of flavi-like viruses found in invertebrates, as well as their deep phylogenetic positions, suggests that they may represent the ancestral forms from which the vertebrate-infecting viruses evolved. For the vertebrate viruses, we expanded the previously mammal-only pegivirus-hepacivirus group to include a virus from the graceful catshark (Proscyllium habereri), which in turn implies that these viruses possess a larger host range than is currently known. In sum, our data show that the Flaviviridae infect a far wider range of hosts and exhibit greater diversity in genome structure than previously anticipated.
Importance: The family Flaviviridae of RNA viruses contains several notorious human pathogens, including dengue virus, West Nile virus, and hepatitis C virus. To date, however, our understanding of the biodiversity and evolution of the Flaviviridae has largely been directed toward vertebrate hosts and their blood-feeding arthropod vectors. Therefore, we investigated an expanded group of potential arthropod and vertebrate host species that have generally been ignored by surveillance programs. Remarkably, these species contained diverse flaviviruses and related viruses that are characterized by major changes in genome size and genome structure, such that these traits are more flexible than previously thought. More generally, these data suggest that arthropods may be the ultimate reservoir of the Flaviviridae and related viruses, harboring considerable genetic and phenotypic diversity. In sum, this study revises the traditional view on the evolutionary history, host range, and genomic structures of a major group of RNA viruses.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
References
-
- Lindenbach BD, Murray CL, Thiel H-J, Rice CM. 2013. Flaviviridae, p 712–746. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Wolters Kluwer, Philadelphia, PA.
-
- Chandriani S, Skewes-Cox P, Zhong W, Ganem DE, Divers TJ, Van Blaricum AJ, Tennant BC, Kistler AL. 2013. Identification of a previously undescribed divergent virus from the Flaviviridae family in an outbreak of equine serum hepatitis. Proc Natl Acad Sci U S A 110:E1407–E1415. doi: 10.1073/pnas.1219217110. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
