

Intracellular Information Processing through Encoding and Decoding of Dynamic Signaling Features
- PMID: 26491963
- PMCID: PMC4619640
- DOI: 10.1371/journal.pcbi.1004563
Intracellular Information Processing through Encoding and Decoding of Dynamic Signaling Features
Abstract
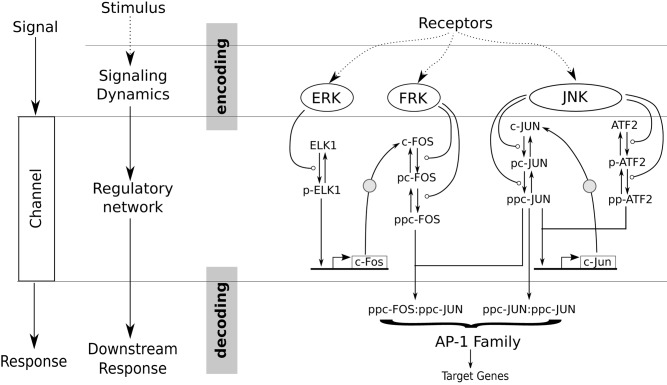
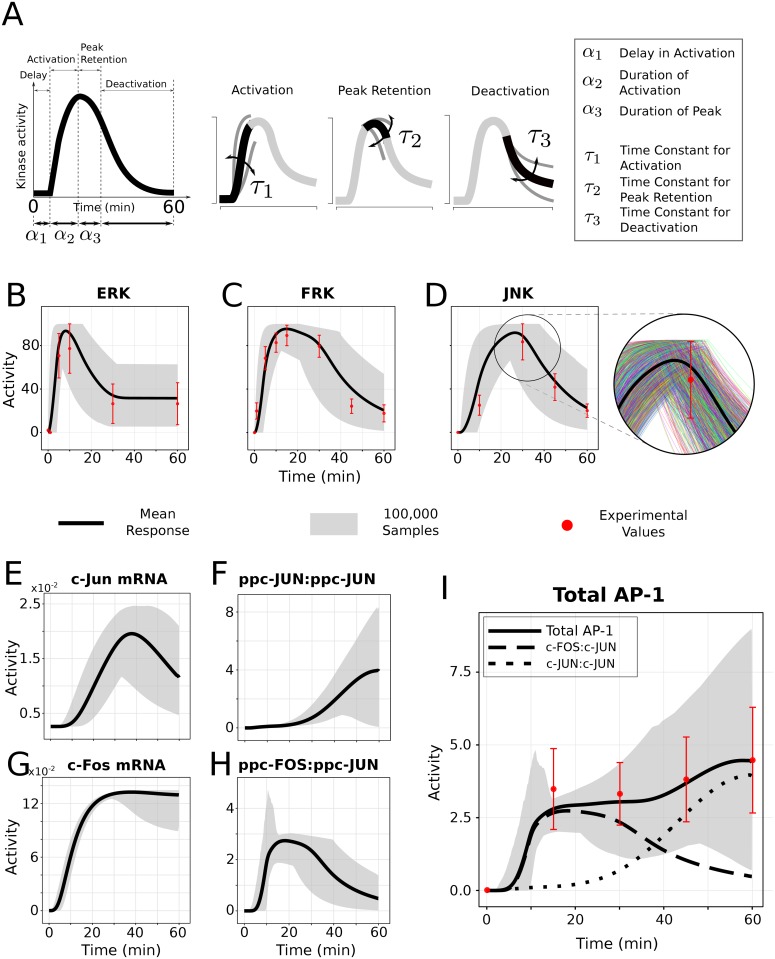
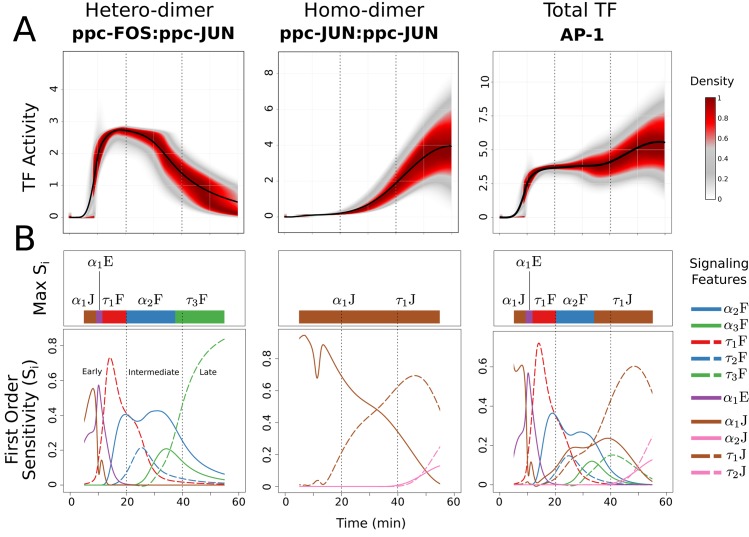
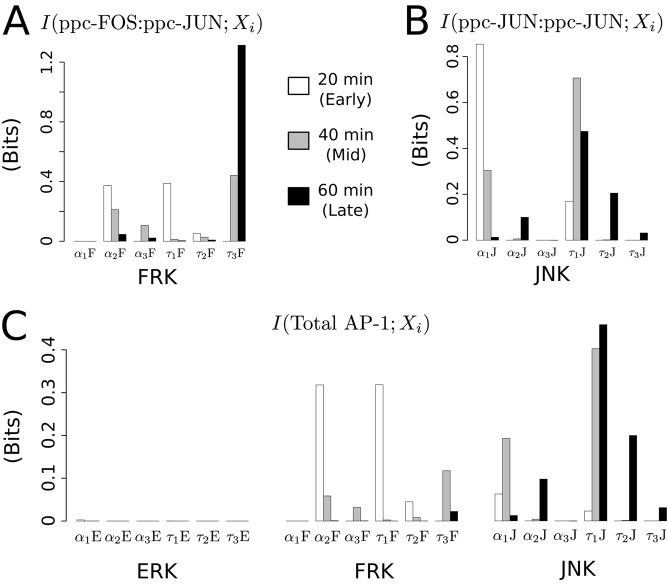
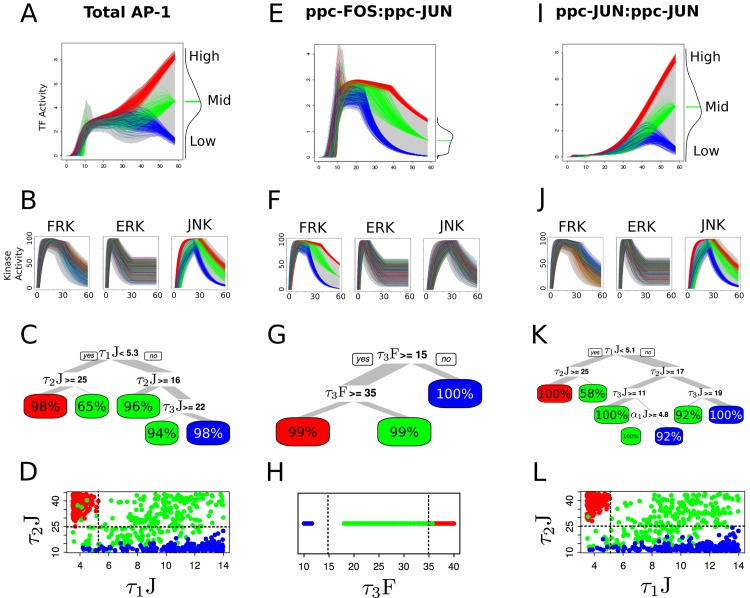
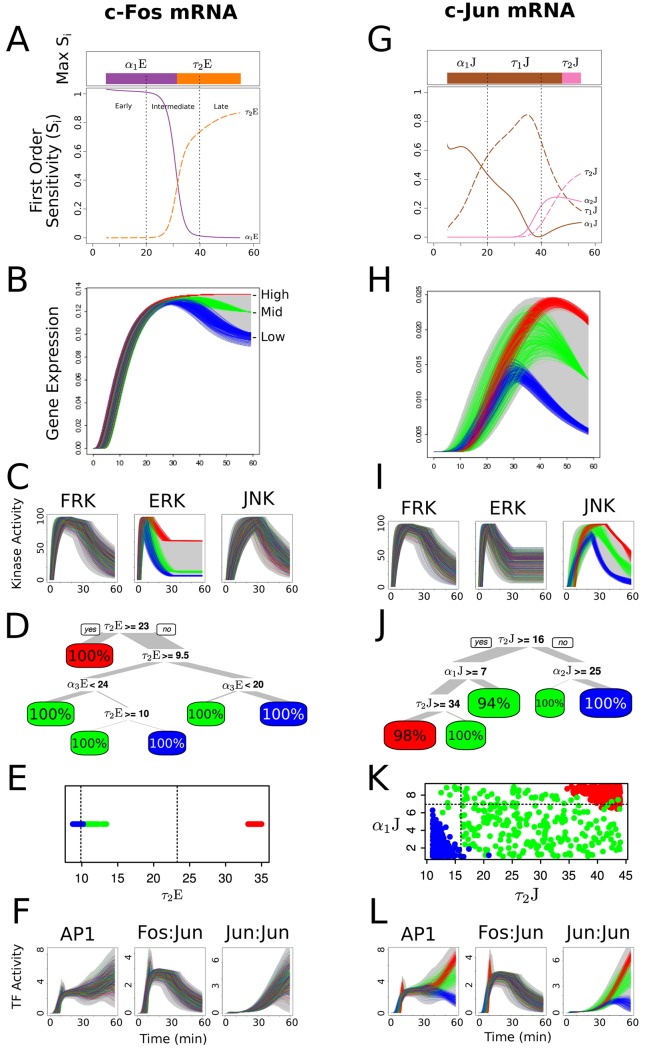
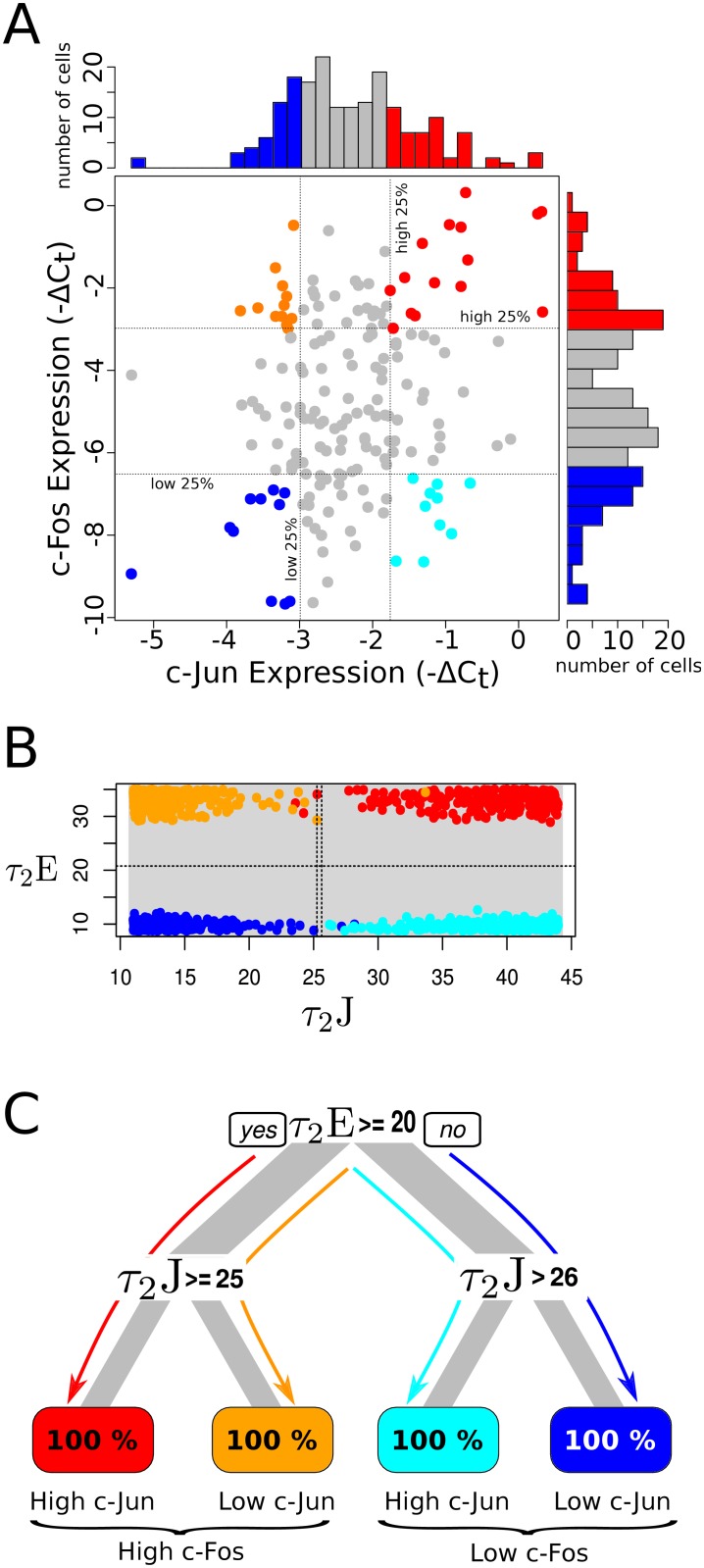
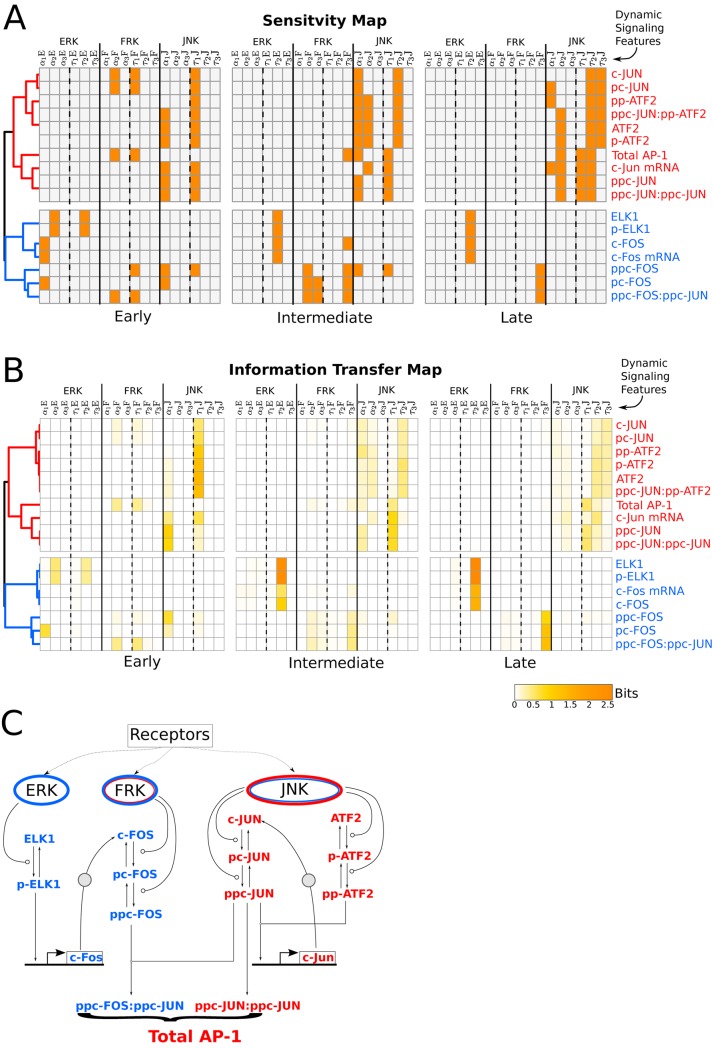
Cell signaling dynamics and transcriptional regulatory activities are variable within specific cell types responding to an identical stimulus. In addition to studying the network interactions, there is much interest in utilizing single cell scale data to elucidate the non-random aspects of the variability involved in cellular decision making. Previous studies have considered the information transfer between the signaling and transcriptional domains based on an instantaneous relationship between the molecular activities. These studies predict a limited binary on/off encoding mechanism which underestimates the complexity of biological information processing, and hence the utility of single cell resolution data. Here we pursue a novel strategy that reformulates the information transfer problem as involving dynamic features of signaling rather than molecular abundances. We pursue a computational approach to test if and how the transcriptional regulatory activity patterns can be informative of the temporal history of signaling. Our analysis reveals (1) the dynamic features of signaling that significantly alter transcriptional regulatory patterns (encoding), and (2) the temporal history of signaling that can be inferred from single cell scale snapshots of transcriptional activity (decoding). Immediate early gene expression patterns were informative of signaling peak retention kinetics, whereas transcription factor activity patterns were informative of activation and deactivation kinetics of signaling. Moreover, the information processing aspects varied across the network, with each component encoding a selective subset of the dynamic signaling features. We developed novel sensitivity and information transfer maps to unravel the dynamic multiplexing of signaling features at each of these network components. Unsupervised clustering of the maps revealed two groups that aligned with network motifs distinguished by transcriptional feedforward vs feedback interactions. Our new computational methodology impacts the single cell scale experiments by identifying downstream snapshot measures required for inferring specific dynamical features of upstream signals involved in the regulation of cellular responses.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Inferring the structures of signaling motifs from paired dynamic traces of single cells.PLoS Comput Biol. 2021 Feb 4;17(2):e1008657. doi: 10.1371/journal.pcbi.1008657. eCollection 2021 Feb. PLoS Comput Biol. 2021. PMID: 33539338 Free PMC article.
-
Quantitative inference of dynamic regulatory pathways via microarray data.BMC Bioinformatics. 2005 Mar 7;6:44. doi: 10.1186/1471-2105-6-44. BMC Bioinformatics. 2005. PMID: 15748298 Free PMC article.
-
Regulatory modules that generate biphasic signal response in biological systems.Syst Biol (Stevenage). 2004 Jun;1(1):139-48. doi: 10.1049/sb:20045014. Syst Biol (Stevenage). 2004. PMID: 17052124
-
Sensing your surroundings: how transcription-regulatory networks of the cell discern environmental signals.Sci STKE. 2005 May 3;2005(282):pe20. doi: 10.1126/stke.2822005pe20. Sci STKE. 2005. PMID: 15870424 Review.
-
[Methods of studying transcriptional regulation of gene expression].Nihon Rinsho. 1996 Feb;54(2):316-21. Nihon Rinsho. 1996. PMID: 8838075 Review. Japanese.
Cited by
-
Network Motifs Capable of Decoding Transcription Factor Dynamics.Sci Rep. 2018 Feb 26;8(1):3594. doi: 10.1038/s41598-018-21945-2. Sci Rep. 2018. PMID: 29483553 Free PMC article.
-
Modeling cytokine regulatory network dynamics driving neuroinflammation in central nervous system disorders.Drug Discov Today Dis Models. 2016 Spring;19:59-67. doi: 10.1016/j.ddmod.2017.01.003. Epub 2017 Apr 10. Drug Discov Today Dis Models. 2016. PMID: 28947907 Free PMC article.
-
Systems analysis of latent HIV reversal reveals altered stress kinase signaling and increased cell death in infected T cells.Sci Rep. 2017 Nov 23;7(1):16179. doi: 10.1038/s41598-017-15532-0. Sci Rep. 2017. PMID: 29170390 Free PMC article.
-
Recognition of sounds by ensembles of proteinoids.Mater Today Bio. 2024 Feb 13;25:100989. doi: 10.1016/j.mtbio.2024.100989. eCollection 2024 Apr. Mater Today Bio. 2024. PMID: 38384791 Free PMC article.
-
On Information Extraction and Decoding Mechanisms Improved by Noisy Amplification in Signaling Pathways.Sci Rep. 2019 Oct 7;9(1):14365. doi: 10.1038/s41598-019-50631-0. Sci Rep. 2019. PMID: 31591406 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
