

Proteasomal inhibition sensitizes cervical cancer cells to mitomycin C-induced bystander effect: the role of tumor microenvironment
- PMID: 26492368
- PMCID: PMC4632313
- DOI: 10.1038/cddis.2015.292
Proteasomal inhibition sensitizes cervical cancer cells to mitomycin C-induced bystander effect: the role of tumor microenvironment
Abstract
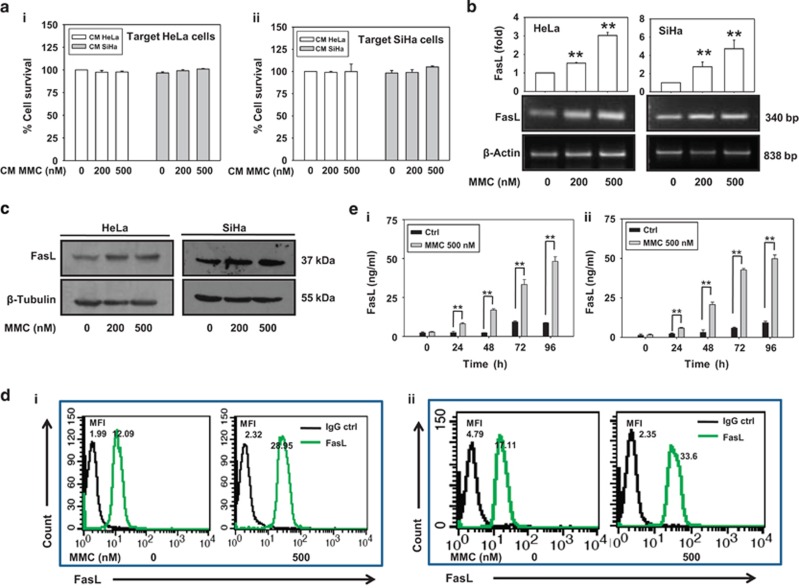
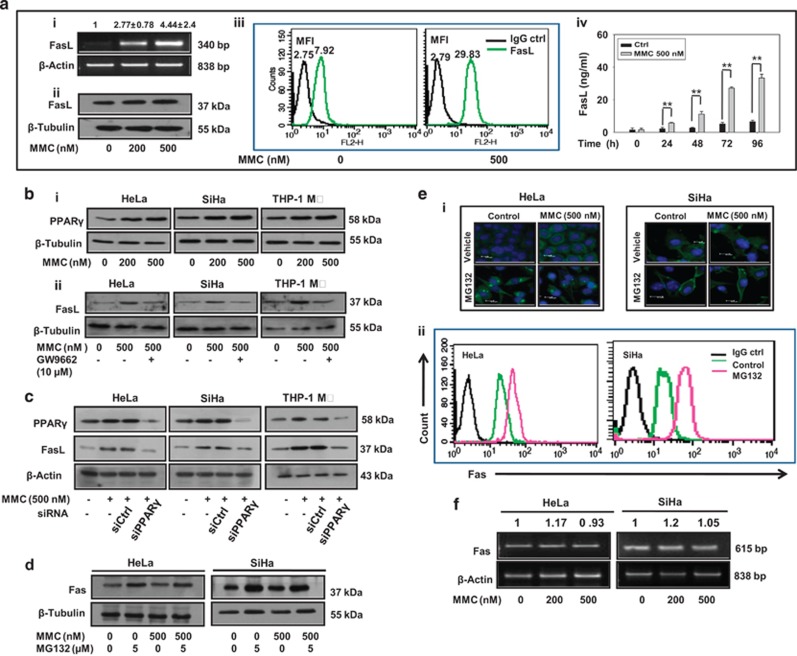
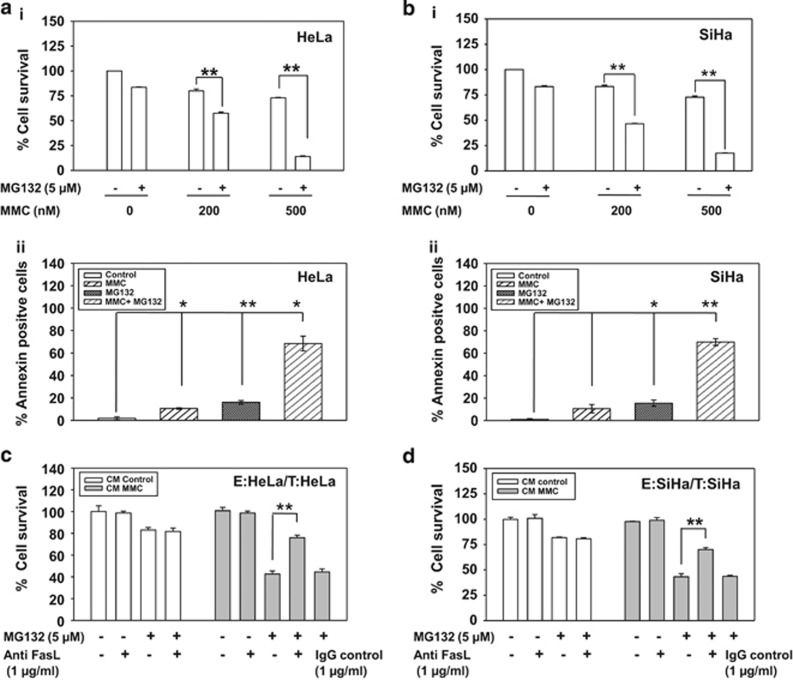
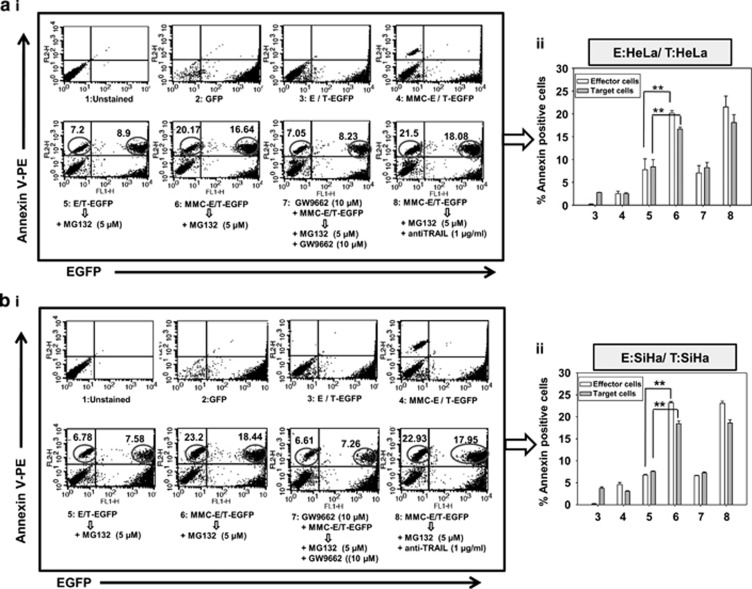
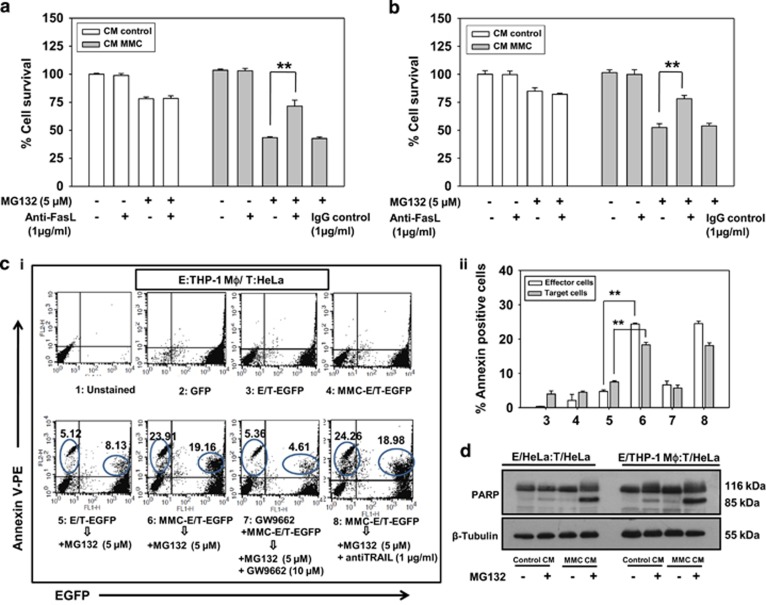
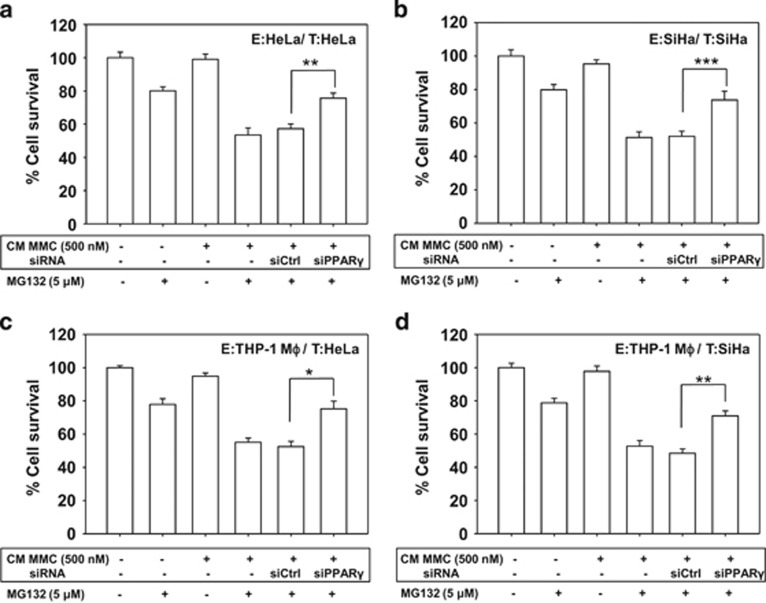
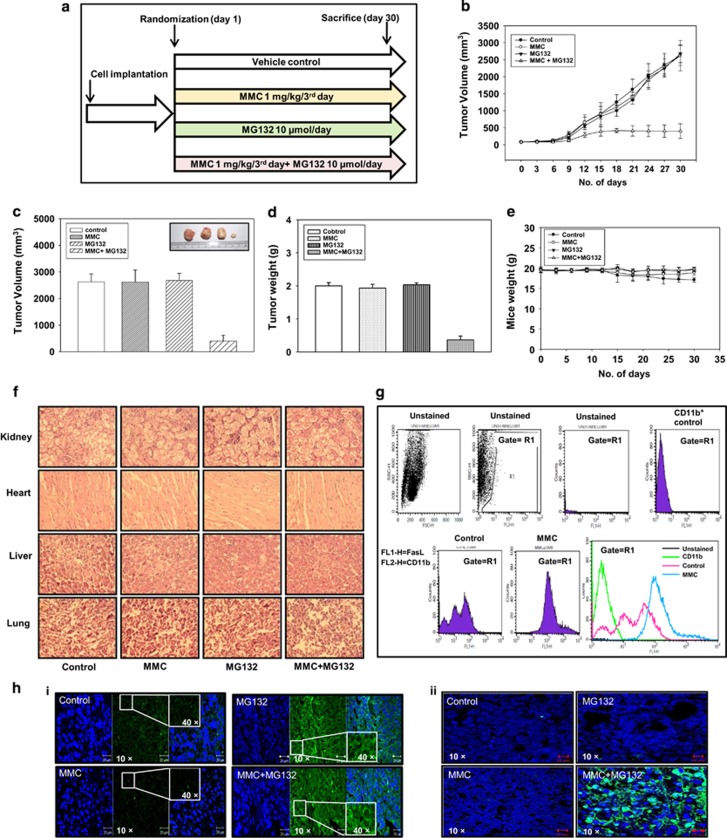
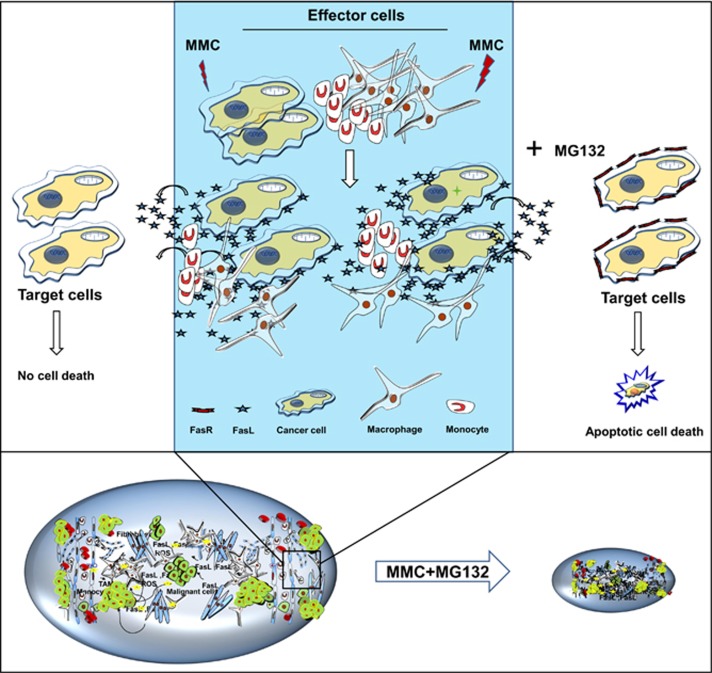
Inaccessibility of drugs to poorly vascularized strata of tumor is one of the limiting factors in cancer therapy. With the advent of bystander effect (BE), it is possible to perpetuate the cellular damage from drug-exposed cells to the unexposed ones. However, the role of infiltrating tumor-associated macrophages (TAMs), an integral part of the tumor microenvironment, in further intensifying BE remains obscure. In the present study, we evaluated the effect of mitomycin C (MMC), a chemotherapeutic drug, to induce BE in cervical carcinoma. By using cervical cancer cells and differentiated macrophages, we demonstrate that MMC induces the expression of FasL via upregulation of PPARγ in both cell types (effector cells) in vitro, but it failed to induce bystander killing in cervical cancer cells. This effect was primarily owing to the proteasomal degradation of death receptors in the cervical cancer cells. Pre-treatment of cervical cancer cells with MG132, a proteasomal inhibitor, facilitates MMC-mediated bystander killing in co-culture and condition medium transfer experiments. In NOD/SCID mice bearing xenografted HeLa tumors administered with the combination of MMC and MG132, tumor progression was significantly reduced in comparison with those treated with either agent alone. FasL expression was increased in TAMs, and the enhanced level of Fas was observed in these tumor sections, thereby causing increased apoptosis. These findings suggest that restoration of death receptor-mediated apoptotic pathway in tumor cells with concomitant activation of TAMs could effectively restrict tumor growth.
Figures
References
-
- 1Mothersill C, Seymour CB. Radiation induced bystander effects—implications for cancer. Nat Rev Cancer 2004; 4: 158–164. - PubMed
-
- 4Ramesh R, Marrogi AJ, Munshi A, Abboud CN, Freeman SM. In vivo analysis of the 'bystander effect': a cytokine cascade. Exp Hematol 1996; 24: 829–838. - PubMed
-
- 5Chhipa RR, Bhat MK. Bystander killing of breast cancer MCF-7 cells by MDA-MB- 231 cells exposed to 5-fluorouracil is mediated via Fas. J Cell Biochem 2007; 101: 68–79. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
