

DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs
- PMID: 26493208
- PMCID: PMC4619085
- DOI: 10.1186/s12864-015-2086-z
DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs
Erratum in
-
Erratum to: 'DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs'.BMC Genomics. 2016 Mar 9;17:215. doi: 10.1186/s12864-016-2544-2. BMC Genomics. 2016. PMID: 26960900 Free PMC article. No abstract available.
Abstract
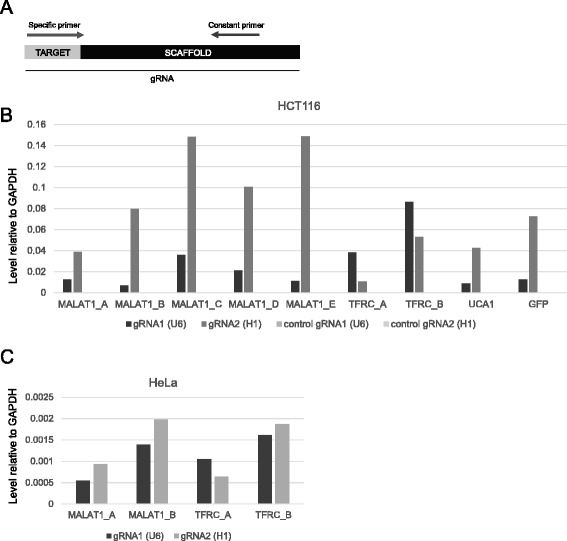
Background: CRISPR genome-editing technology makes it possible to quickly and cheaply delete non-protein-coding regulatory elements. We present a vector system adapted for this purpose called DECKO (Double Excision CRISPR Knockout), which applies a simple two-step cloning to generate lentiviral vectors expressing two guide RNAs (gRNAs) simultaneously. The key feature of DECKO is its use of a single 165 bp starting oligonucleotide carrying the variable sequences of both gRNAs, making it fully scalable from single-locus studies to complex library cloning.
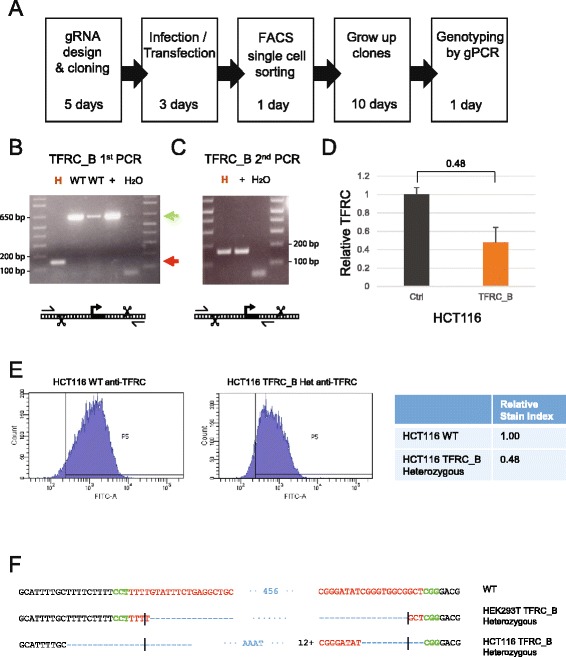
Results: We apply DECKO to deleting the promoters of one protein-coding gene and two oncogenic lncRNAs, UCA1 and the highly-expressed MALAT1, focus of many previous studies employing RNA interference approaches. DECKO successfully deleted genomic fragments ranging in size from 100 to 3000 bp in four human cell lines. Using a clone-derivation workflow lasting approximately 20 days, we obtained 9 homozygous and 17 heterozygous promoter knockouts in three human cell lines. Frequent target region inversions were observed. These clones have reductions in steady-state MALAT1 RNA levels of up to 98 % and display reduced proliferation rates.
Conclusions: We present a dual CRISPR tool, DECKO, which is cloned using a single starting oligonucleotide, thereby affording simplicity and scalability to CRISPR knockout studies of non-coding genomic elements, including long non-coding RNAs.
Figures




Similar articles
-
Deletion of Regulatory Elements with All-in-One CRISPR-Cas9 Vectors.Methods Mol Biol. 2021;2351:321-334. doi: 10.1007/978-1-0716-1597-3_18. Methods Mol Biol. 2021. PMID: 34382198
-
Production and Validation of Lentiviral Vectors for CRISPR/Cas9 Delivery.Methods Mol Biol. 2019;1961:93-109. doi: 10.1007/978-1-4939-9170-9_7. Methods Mol Biol. 2019. PMID: 30912042
-
Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library.Nat Biotechnol. 2014 Mar;32(3):267-73. doi: 10.1038/nbt.2800. Epub 2013 Dec 23. Nat Biotechnol. 2014. PMID: 24535568
-
Designing libraries for pooled CRISPR functional screens of long noncoding RNAs.Mamm Genome. 2022 Jun;33(2):312-327. doi: 10.1007/s00335-021-09918-9. Epub 2021 Sep 17. Mamm Genome. 2022. PMID: 34533605 Free PMC article. Review.
-
The applications of CRISPR/Cas-mediated microRNA and lncRNA editing in plant biology: shaping the future of plant non-coding RNA research.Planta. 2023 Dec 28;259(2):32. doi: 10.1007/s00425-023-04303-z. Planta. 2023. PMID: 38153530 Review.
Cited by
-
Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing.Genome Biol. 2018 Oct 19;19(1):170. doi: 10.1186/s13059-018-1518-x. Genome Biol. 2018. PMID: 30340517 Free PMC article.
-
Alternative splicing modulation mediated by G-quadruplex structures in MALAT1 lncRNA.Nucleic Acids Res. 2022 Jan 11;50(1):378-396. doi: 10.1093/nar/gkab1066. Nucleic Acids Res. 2022. Retraction in: Nucleic Acids Res. 2023 Sep 22;51(17):9508. doi: 10.1093/nar/gkad654. PMID: 34761272 Free PMC article. Retracted.
-
LncRNAs in Cancer: From garbage to Junk.Cancers (Basel). 2020 Oct 31;12(11):3220. doi: 10.3390/cancers12113220. Cancers (Basel). 2020. PMID: 33142861 Free PMC article. Review.
-
Parallelized engineering of mutational models using piggyBac transposon delivery of CRISPR libraries.Cell Rep Methods. 2024 Jan 22;4(1):100672. doi: 10.1016/j.crmeth.2023.100672. Epub 2023 Dec 12. Cell Rep Methods. 2024. PMID: 38091988 Free PMC article.
-
Recent advances in functional genome analysis.F1000Res. 2018 Dec 21;7:F1000 Faculty Rev-1968. doi: 10.12688/f1000research.15274.1. eCollection 2018. F1000Res. 2018. PMID: 30613379 Free PMC article. Review.
References
-
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89. doi: 10.1101/gr.132159.111. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
