

ARK: Aggregation of Reads by K-Means for Estimation of Bacterial Community Composition
- PMID: 26496191
- PMCID: PMC4619776
- DOI: 10.1371/journal.pone.0140644
ARK: Aggregation of Reads by K-Means for Estimation of Bacterial Community Composition
Abstract
Motivation: Estimation of bacterial community composition from high-throughput sequenced 16S rRNA gene amplicons is a key task in microbial ecology. Since the sequence data from each sample typically consist of a large number of reads and are adversely impacted by different levels of biological and technical noise, accurate analysis of such large datasets is challenging.
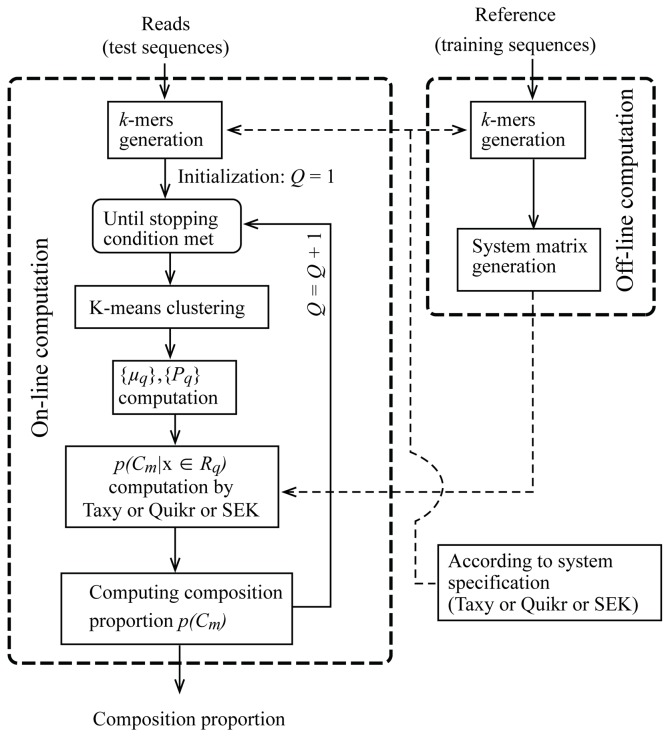
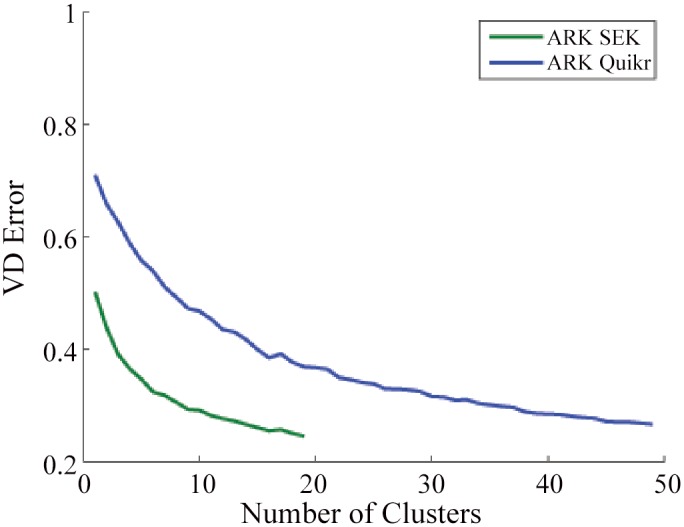
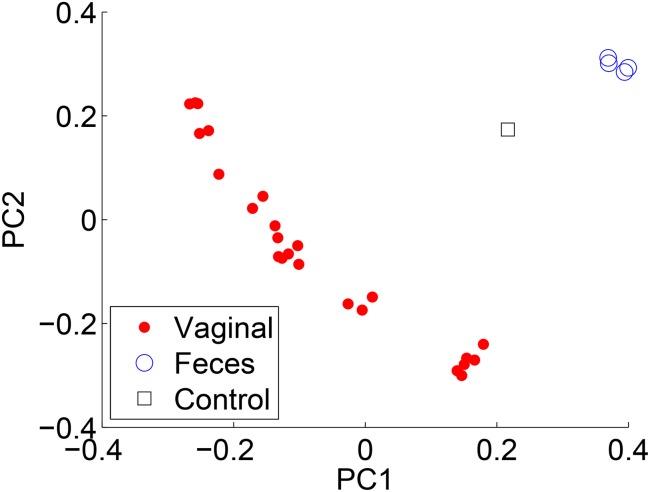
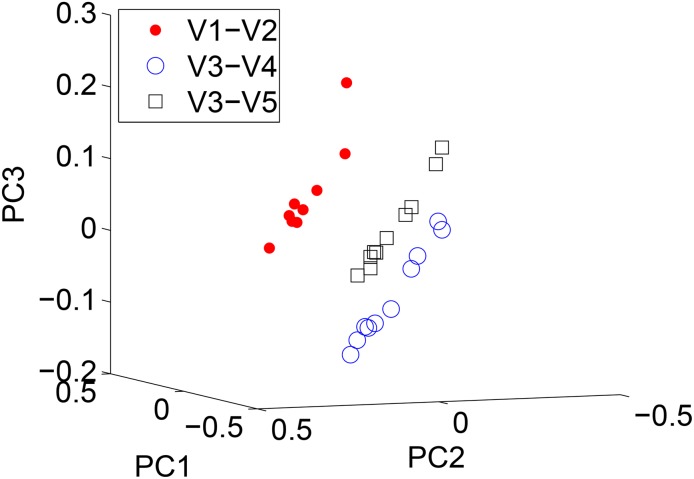
Results: There has been a recent surge of interest in using compressed sensing inspired and convex-optimization based methods to solve the estimation problem for bacterial community composition. These methods typically rely on summarizing the sequence data by frequencies of low-order k-mers and matching this information statistically with a taxonomically structured database. Here we show that the accuracy of the resulting community composition estimates can be substantially improved by aggregating the reads from a sample with an unsupervised machine learning approach prior to the estimation phase. The aggregation of reads is a pre-processing approach where we use a standard K-means clustering algorithm that partitions a large set of reads into subsets with reasonable computational cost to provide several vectors of first order statistics instead of only single statistical summarization in terms of k-mer frequencies. The output of the clustering is then processed further to obtain the final estimate for each sample. The resulting method is called Aggregation of Reads by K-means (ARK), and it is based on a statistical argument via mixture density formulation. ARK is found to improve the fidelity and robustness of several recently introduced methods, with only a modest increase in computational complexity.
Availability: An open source, platform-independent implementation of the method in the Julia programming language is freely available at https://github.com/dkoslicki/ARK. A Matlab implementation is available at http://www.ee.kth.se/ctsoftware.
Conflict of interest statement
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
