

Early divergent strains of Yersinia pestis in Eurasia 5,000 years ago
- PMID: 26496604
- PMCID: PMC4644222
- DOI: 10.1016/j.cell.2015.10.009
Early divergent strains of Yersinia pestis in Eurasia 5,000 years ago
Abstract
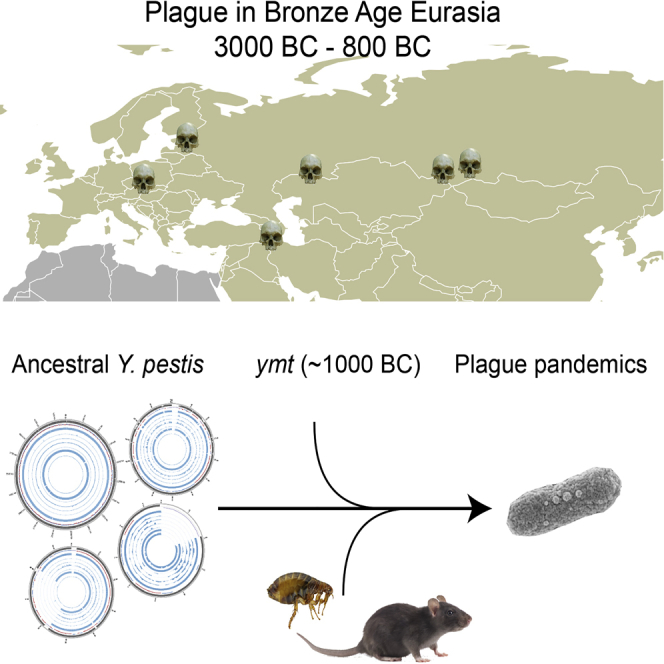
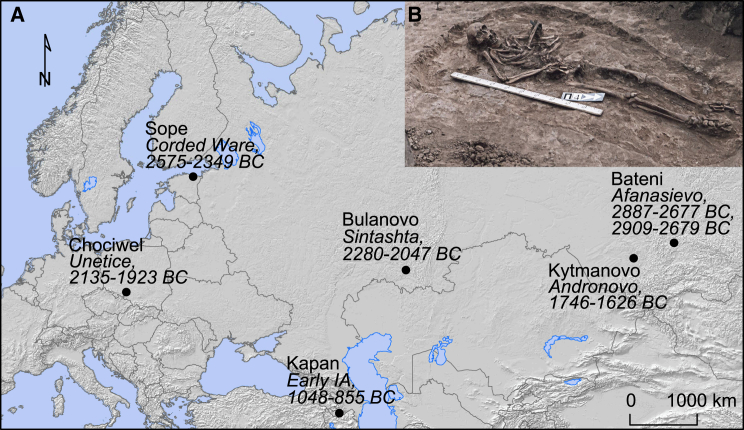
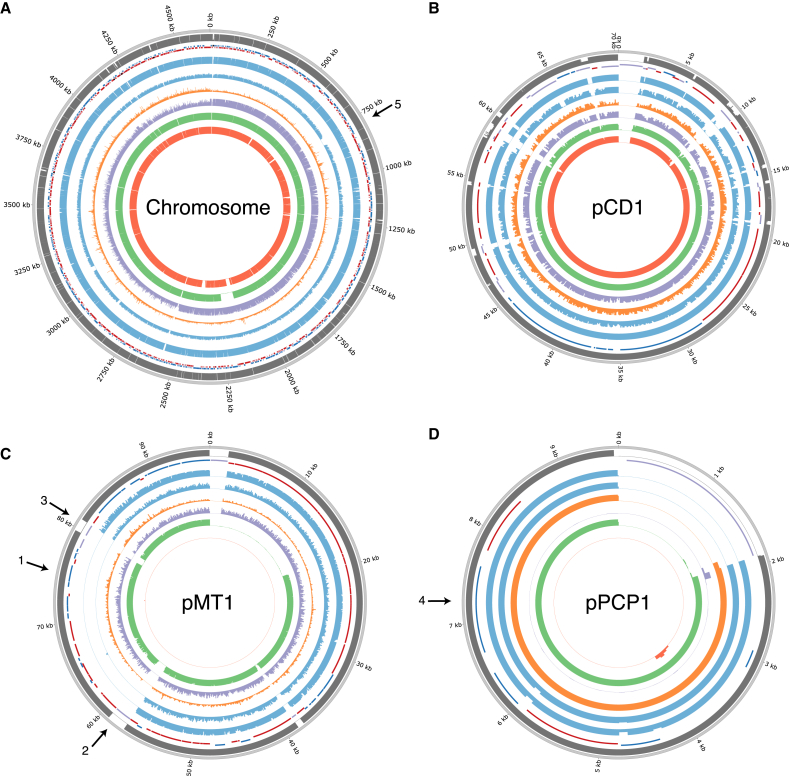
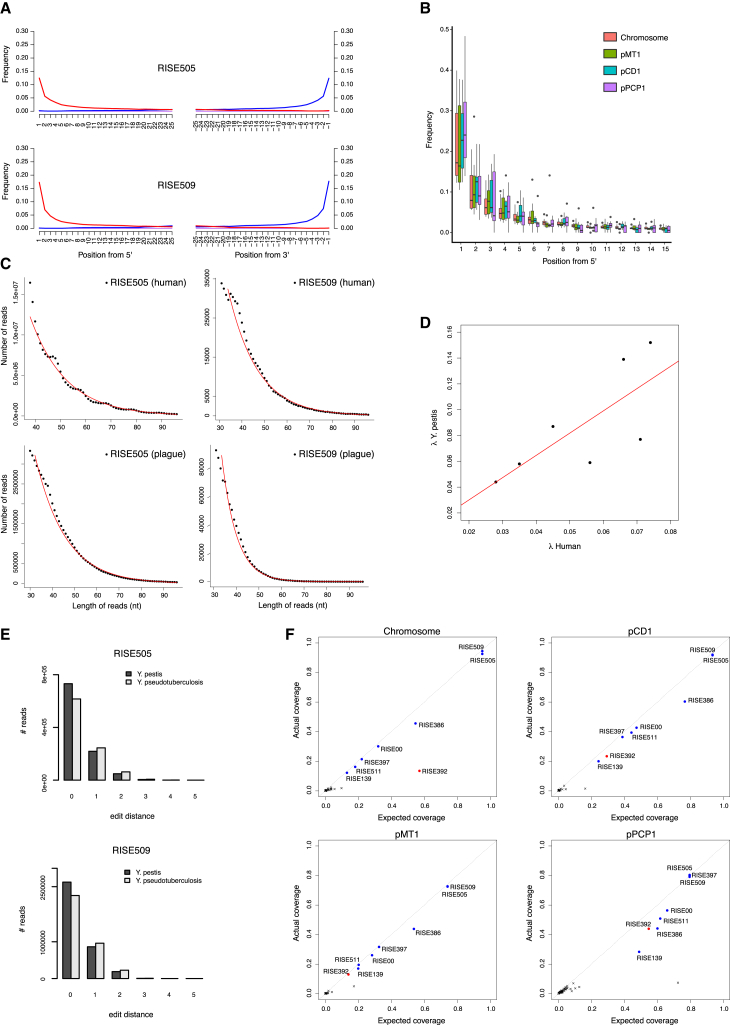
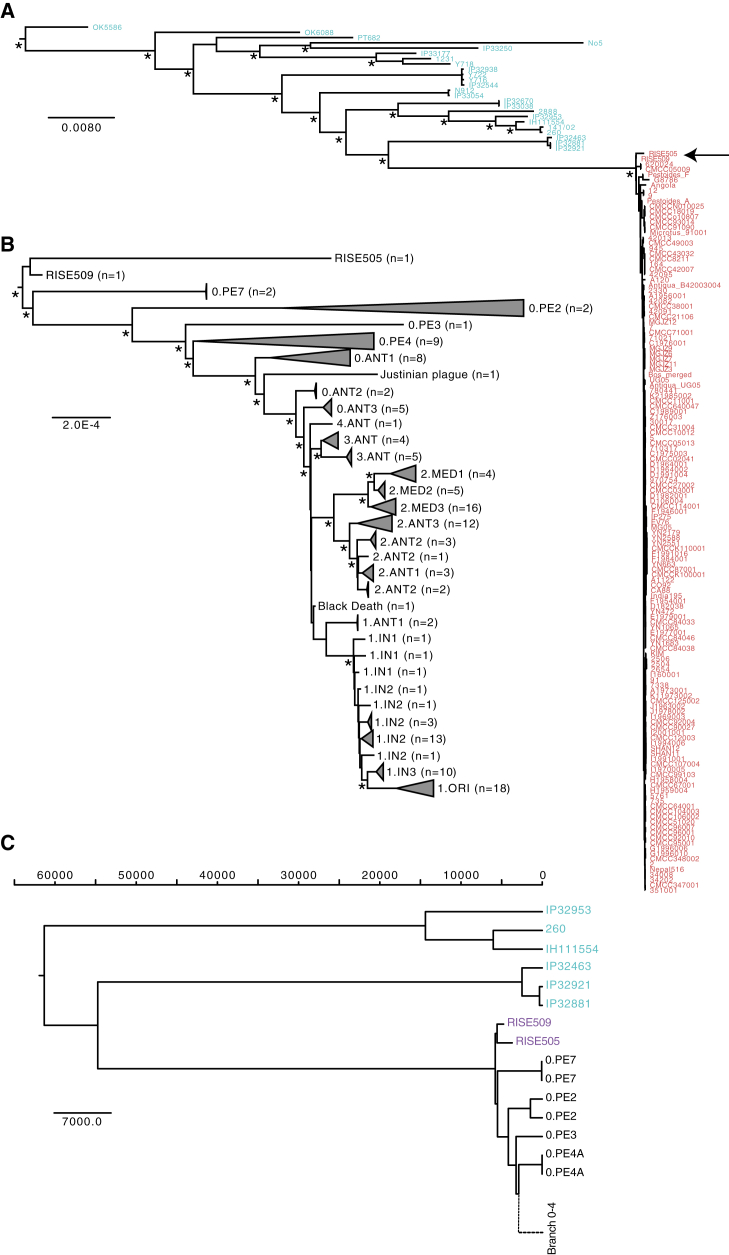
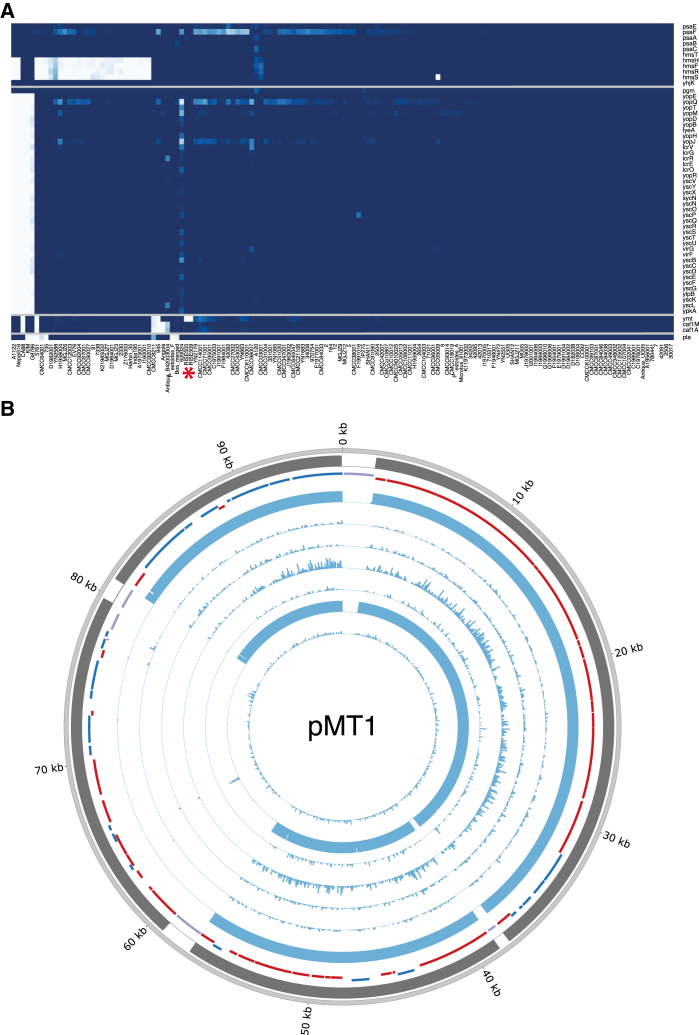
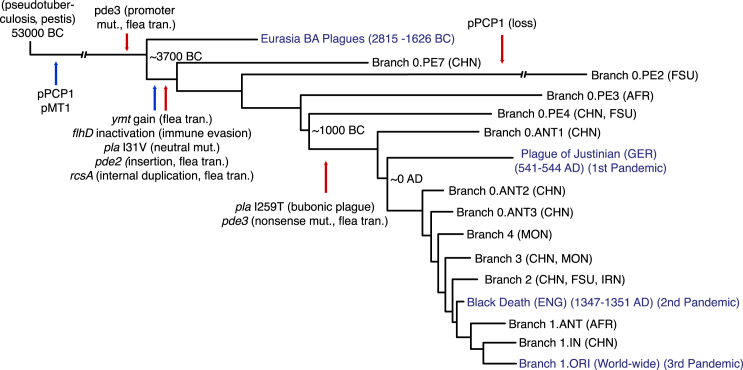
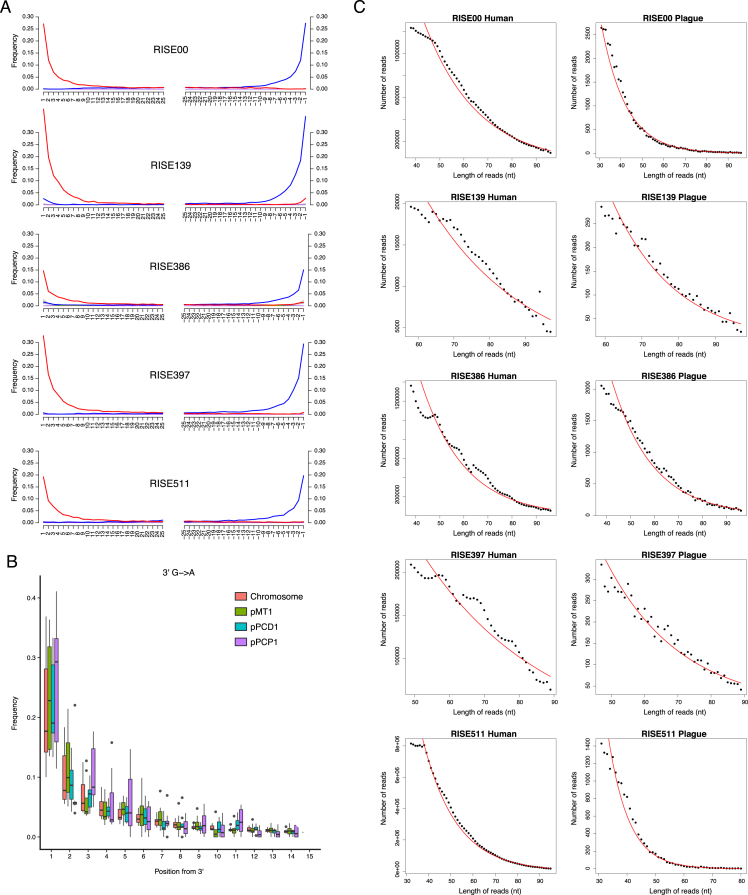
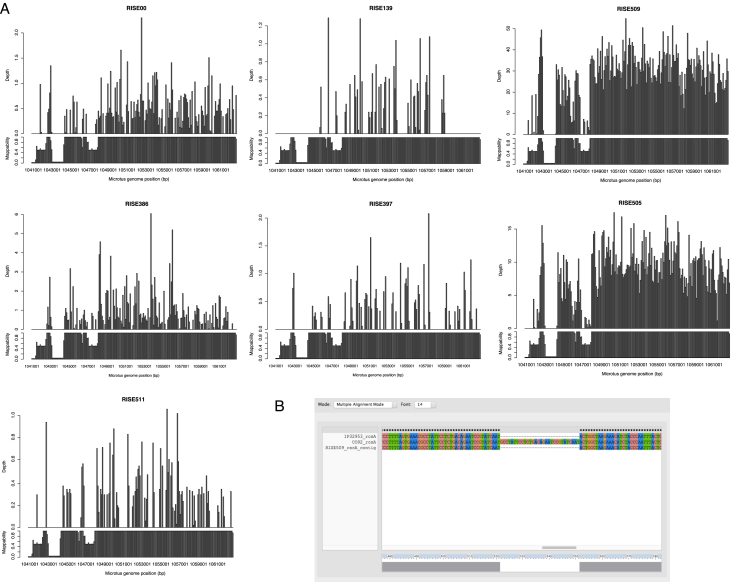
The bacteria Yersinia pestis is the etiological agent of plague and has caused human pandemics with millions of deaths in historic times. How and when it originated remains contentious. Here, we report the oldest direct evidence of Yersinia pestis identified by ancient DNA in human teeth from Asia and Europe dating from 2,800 to 5,000 years ago. By sequencing the genomes, we find that these ancient plague strains are basal to all known Yersinia pestis. We find the origins of the Yersinia pestis lineage to be at least two times older than previous estimates. We also identify a temporal sequence of genetic changes that lead to increased virulence and the emergence of the bubonic plague. Our results show that plague infection was endemic in the human populations of Eurasia at least 3,000 years before any historical recordings of pandemics.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
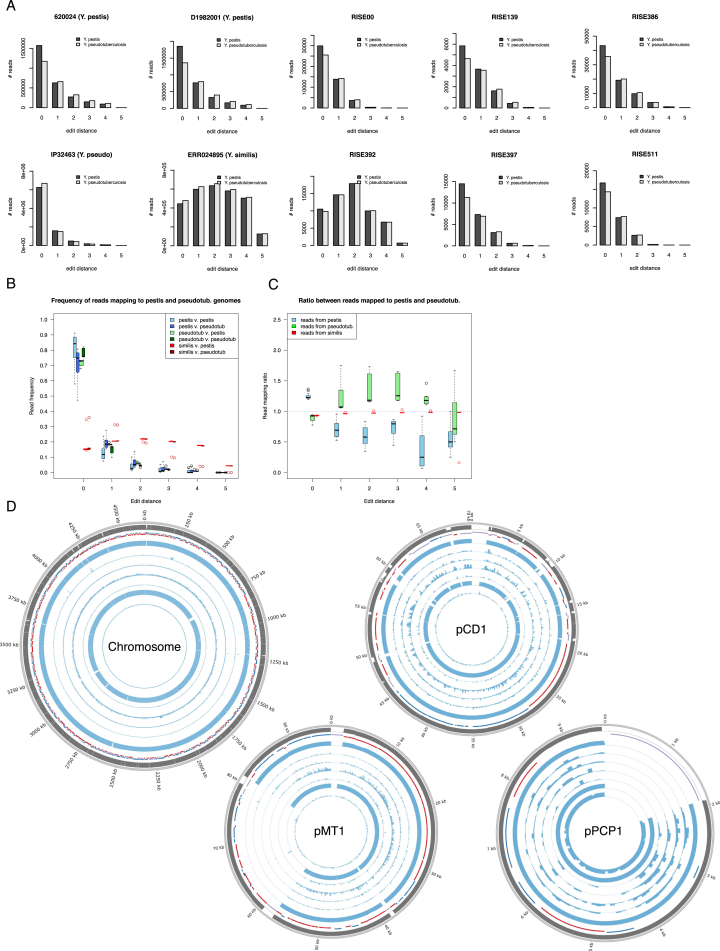
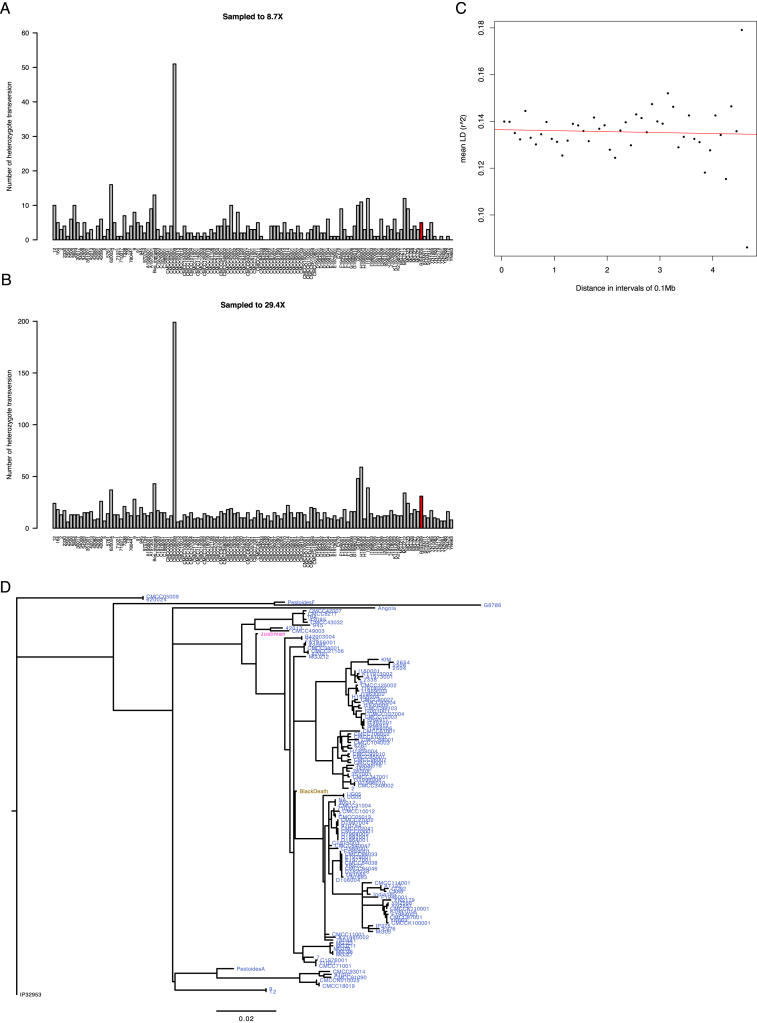
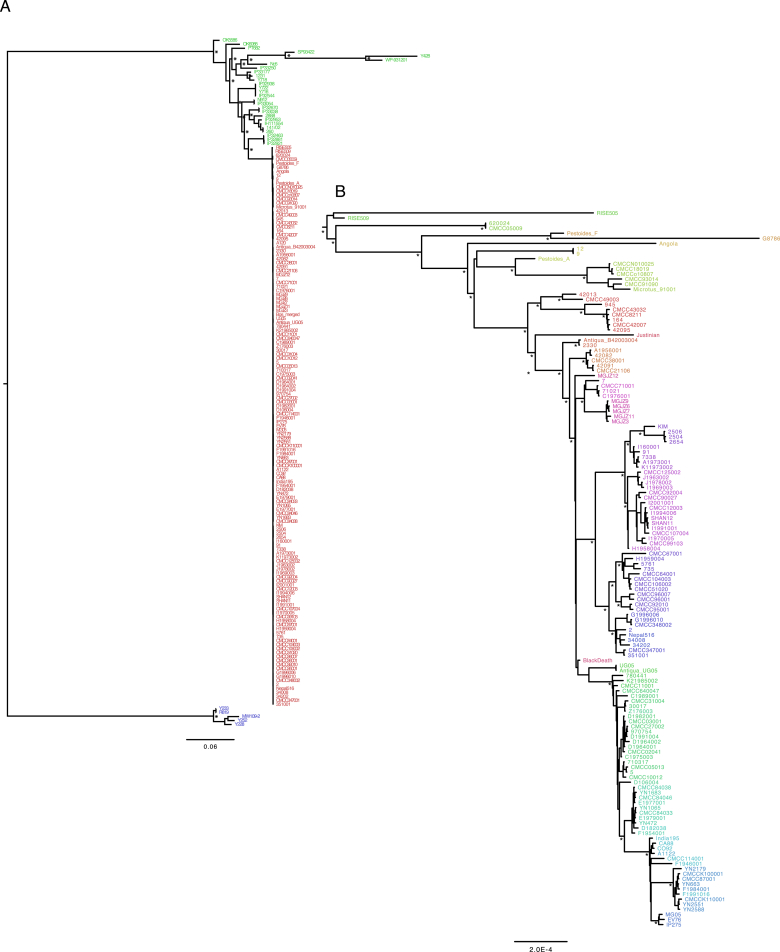
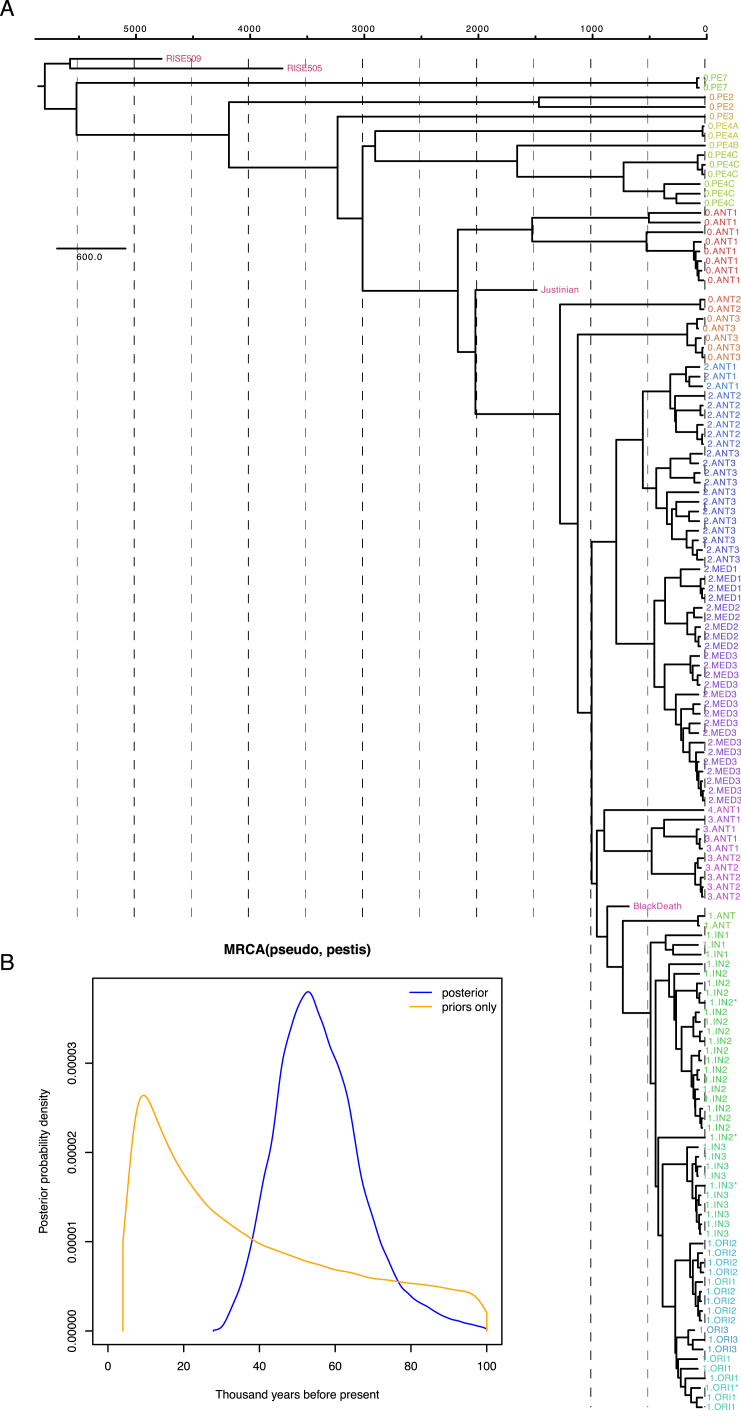
References
-
- Allentoft M.E., Sikora M., Sjögren K.-G., Rasmussen S., Rasmussen M., Stenderup J., Damgaard P.B., Schroeder H., Ahlström T., Vinner L. Population genomics of Bronze Age Eurasia. Nature. 2015;522:167–172. - PubMed
-
- Anthony D. Princeton University Press; Princeton: 2007. The Horse, The Wheel and Language. How Bronze-Age riders from the Eurasian Steppes Shaped the Modern World.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
