

Inhibition of DYRK1A and GSK3B induces human β-cell proliferation
- PMID: 26496802
- PMCID: PMC4639830
- DOI: 10.1038/ncomms9372
Inhibition of DYRK1A and GSK3B induces human β-cell proliferation
Abstract
Insufficient pancreatic β-cell mass or function results in diabetes mellitus. While significant progress has been made in regulating insulin secretion from β-cells in diabetic patients, no pharmacological agents have been described that increase β-cell replication in humans. Here we report aminopyrazine compounds that stimulate robust β-cell proliferation in adult primary islets, most likely as a result of combined inhibition of DYRK1A and GSK3B. Aminopyrazine-treated human islets retain functionality in vitro and after transplantation into diabetic mice. Oral dosing of these compounds in diabetic mice induces β-cell proliferation, increases β-cell mass and insulin content, and improves glycaemic control. Biochemical, genetic and cell biology data point to Dyrk1a as the key molecular target. This study supports the feasibility of treating diabetes with an oral therapy to restore β-cell mass, and highlights a tractable pathway for future drug discovery efforts.
Conflict of interest statement
B.T., Q.J., V.N.-T., A.P., Y.-Q.Z., C.M.F., J.L., L.W., B.B., G.H., M.Q., Y.L., X.H., T.M., S.Y., J.L., T.W., P.M., R.G. and B.L. are employees of the Genomics Institute of the Novartis Research Foundation. A.H. is an employee of Genentech, Inc. W.S. is an employee of the California Institute for Biomedical Research. H.M.S. is an employee of the Novartis Institutes for BioMedical Research. B.H. serves as a consultant for Janssen Pharmaceuticals, Novartis Pharmaceuticals Corporation, and Sanofi-Aventis Group.
Figures
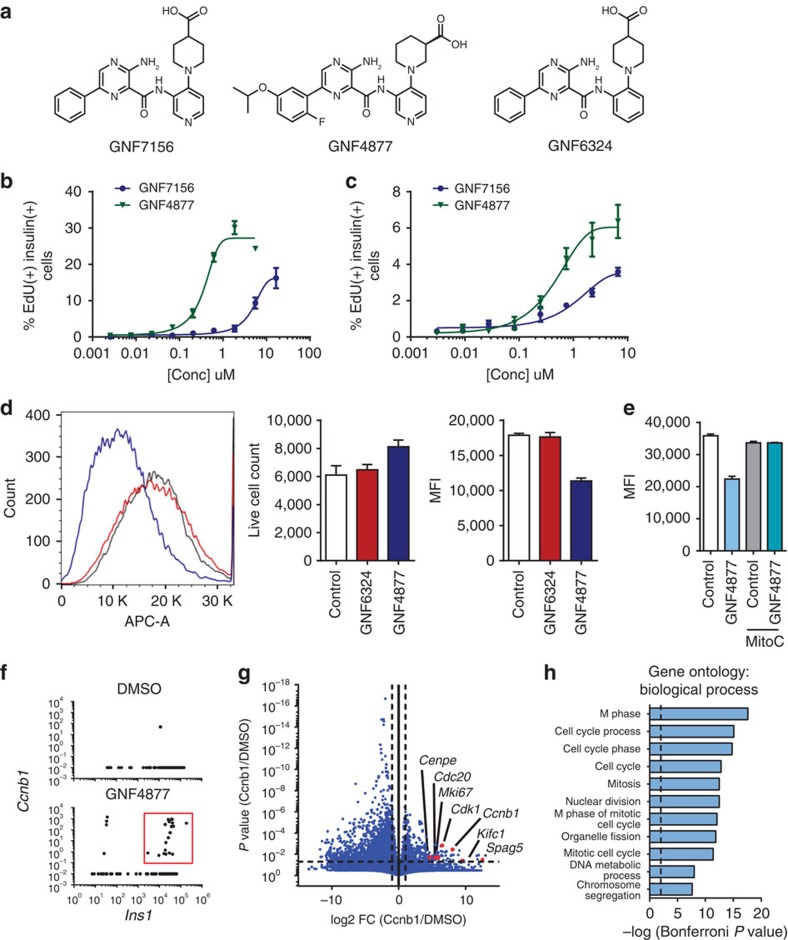
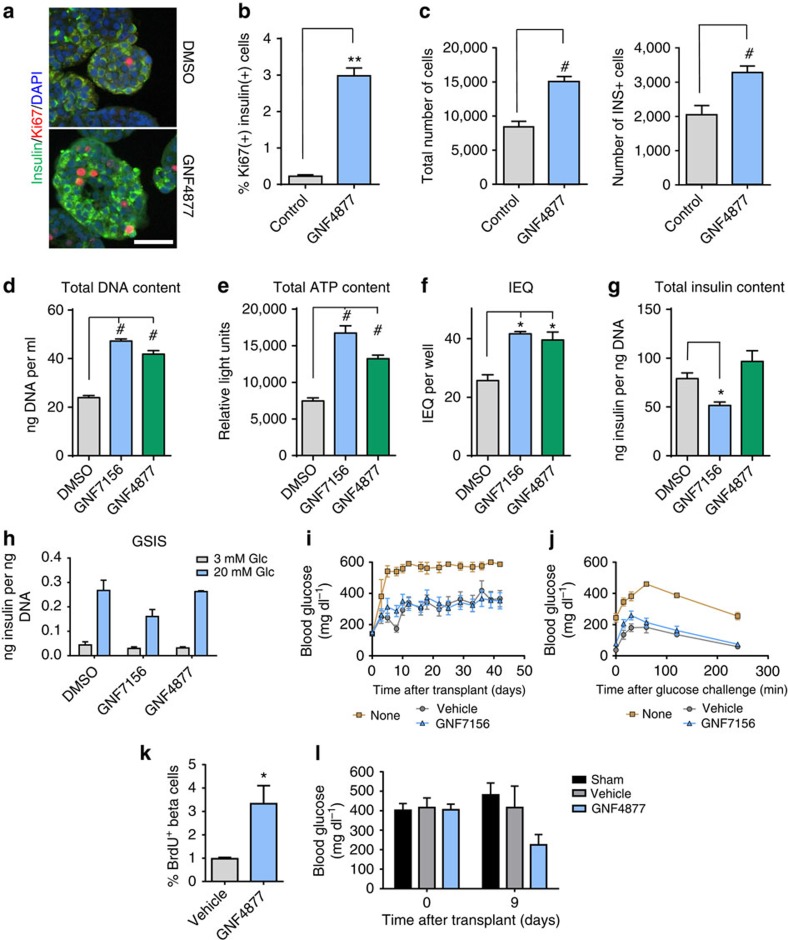
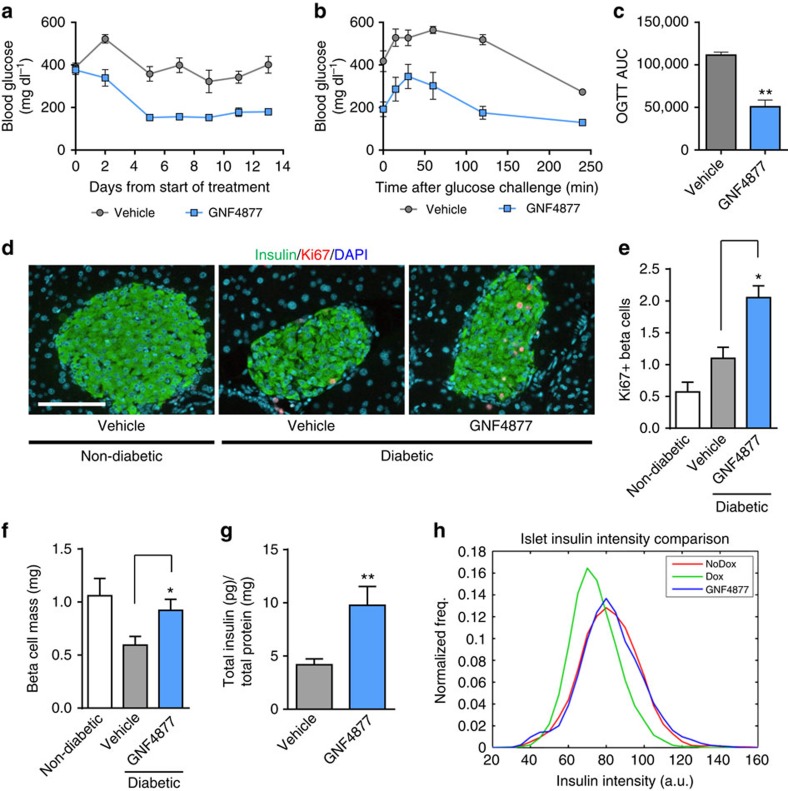
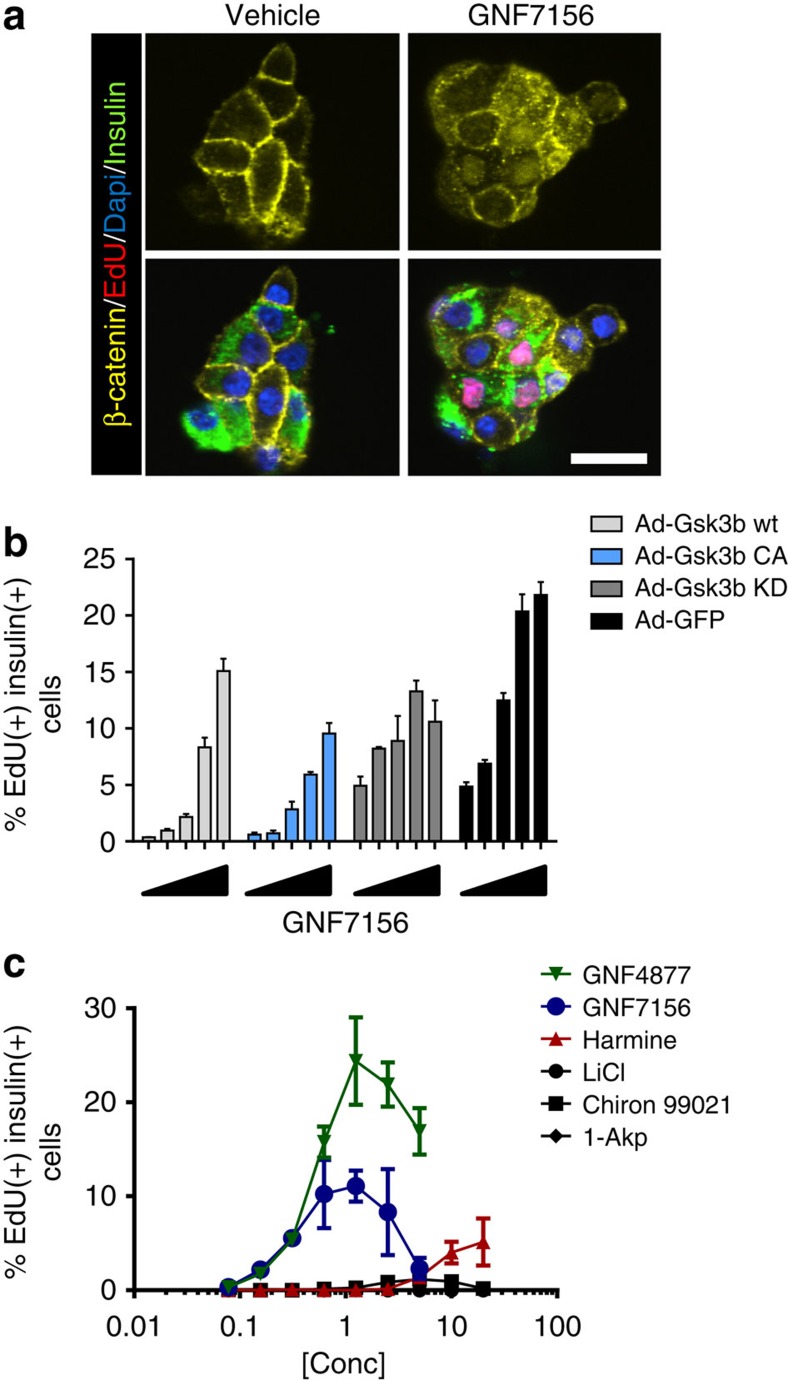
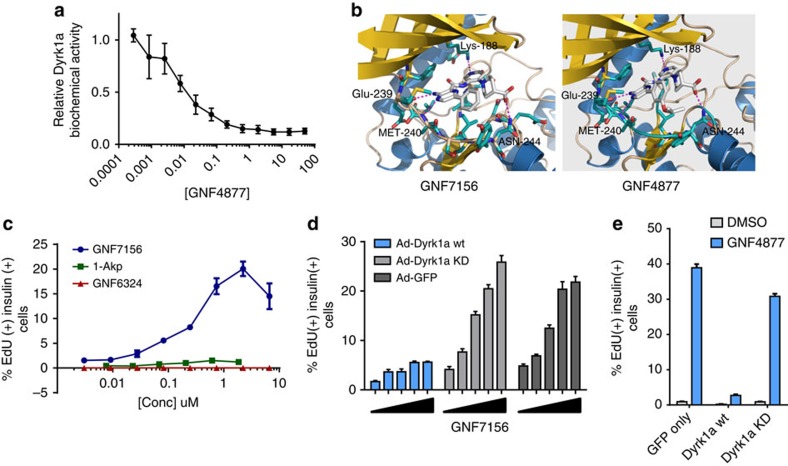
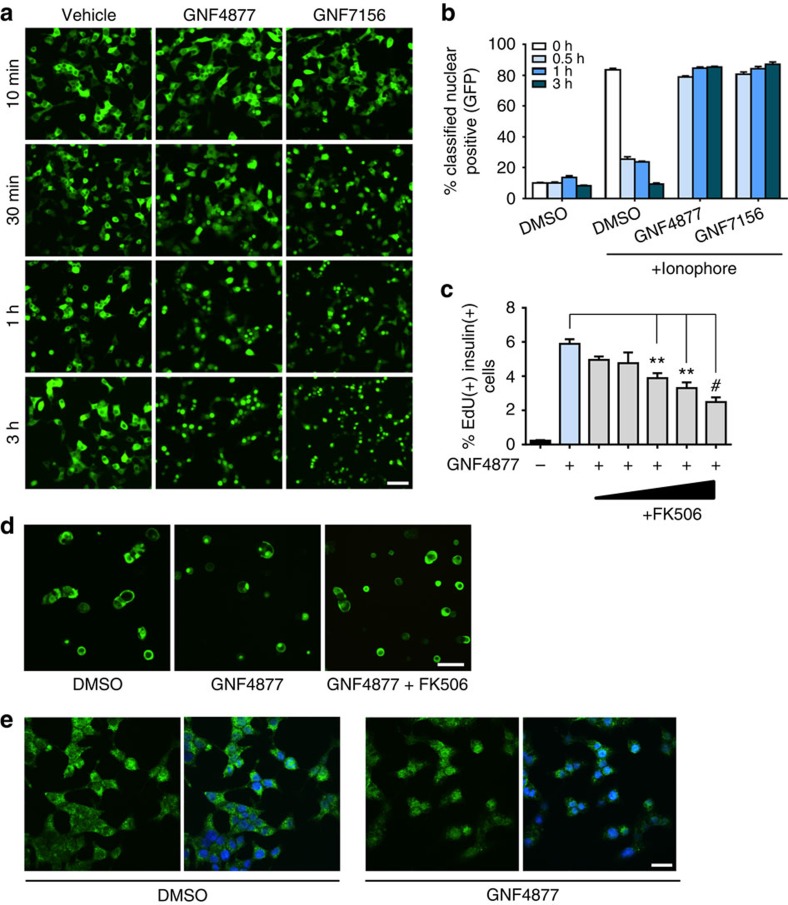
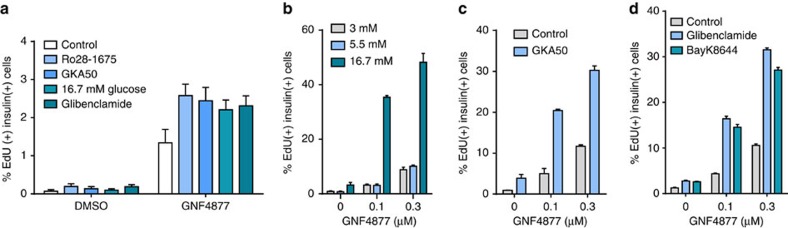
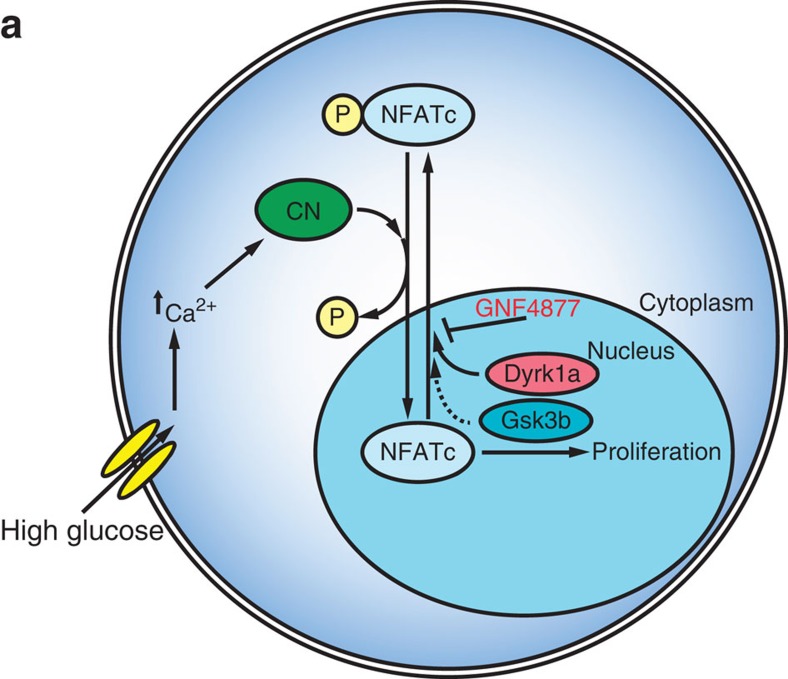
References
-
- Butler A. E. et al.. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110 (2003). - PubMed
-
- Stumvoll M., Goldstein B. J. & van Haeften T. W. Type 2 diabetes: pathogenesis and treatment. Lancet 371, 2153–2156 (2008). - PubMed
-
- Schafer S. A., Machicao F., Fritsche A., Haring H. U. & Kantartzis K. New type 2 diabetes risk genes provide new insights in insulin secretion mechanisms. Diabetes Res. Clin. Pract. 93, (Suppl 1): S9–S24 (2011). - PubMed
-
- Dor Y., Brown J., Martinez O. I. & Melton D. A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46 (2004). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
