

Activin and TGFβ use diverging mitogenic signaling in advanced colon cancer
- PMID: 26497569
- PMCID: PMC4619565
- DOI: 10.1186/s12943-015-0456-4
Activin and TGFβ use diverging mitogenic signaling in advanced colon cancer
Abstract
Background: Understanding cell signaling pathways that contribute to metastatic colon cancer is critical to risk stratification in the era of personalized therapeutics. Here, we dissect the unique involvement of mitogenic pathways in a TGFβ or activin-induced metastatic phenotype of colon cancer.
Method: Mitogenic signaling/growth factor receptor status and p21 localization were correlated in primary colon cancers and intestinal tumors from either AOM/DSS treated ACVR2A (activin receptor 2) -/- or wild type mice. Colon cancer cell lines (+/- SMAD4) were interrogated for ligand-induced PI3K and MEK/ERK pathway activation and downstream protein/phospho-isoform expression/association after knockdown and pharmacologic inhibition of pathway members. EMT was assessed using epithelial/mesenchymal markers and migration assays.
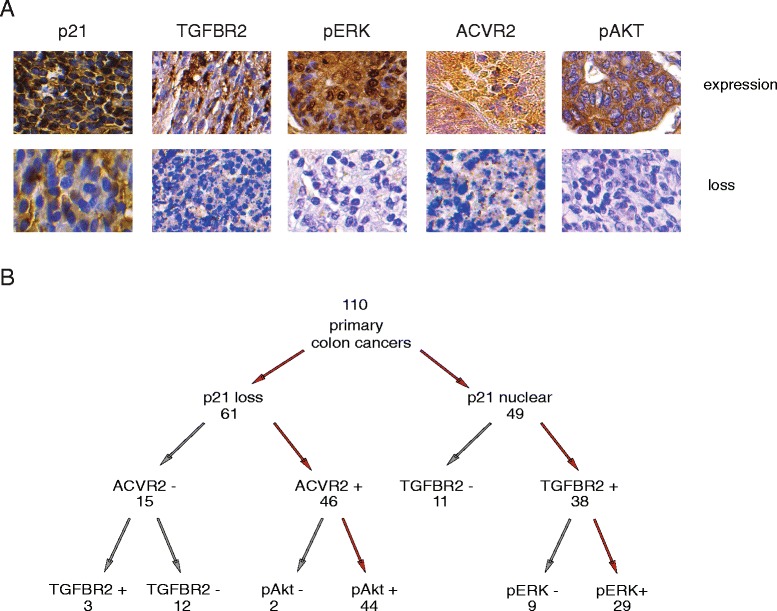
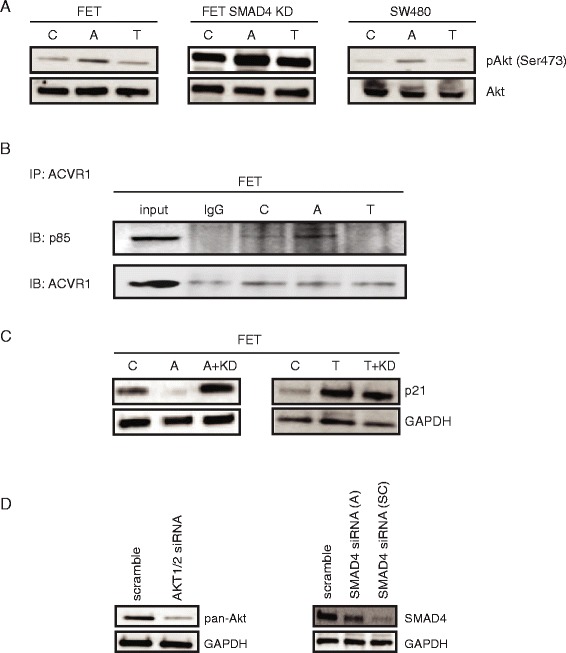
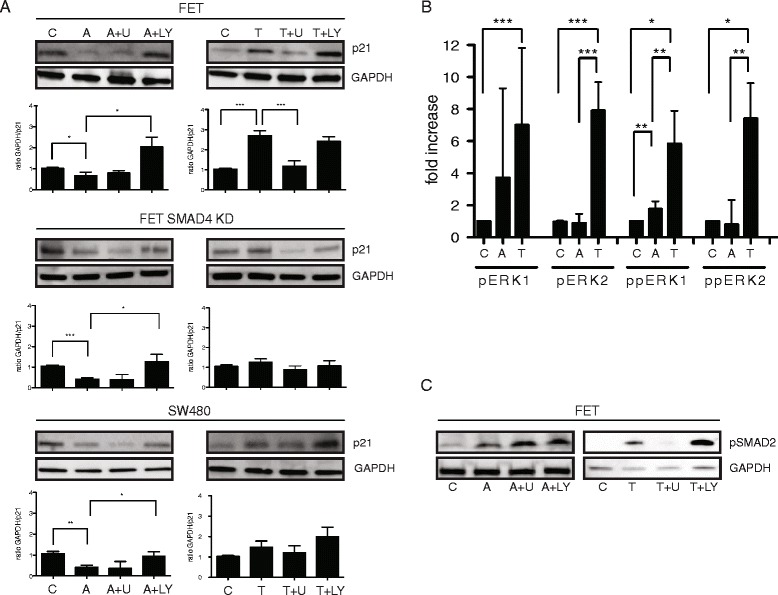
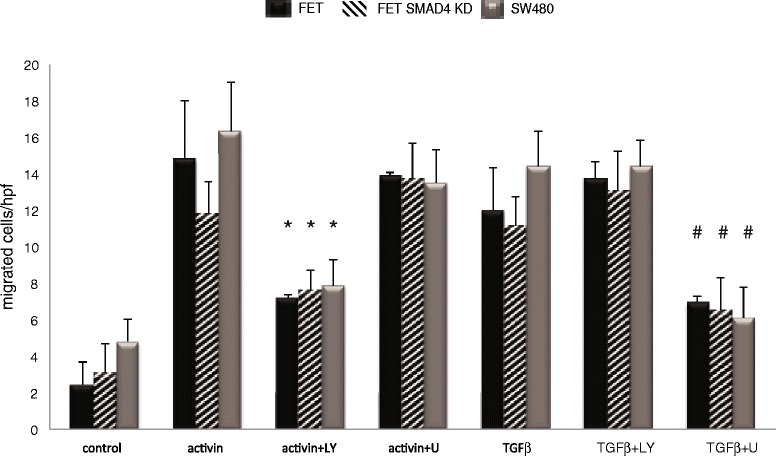
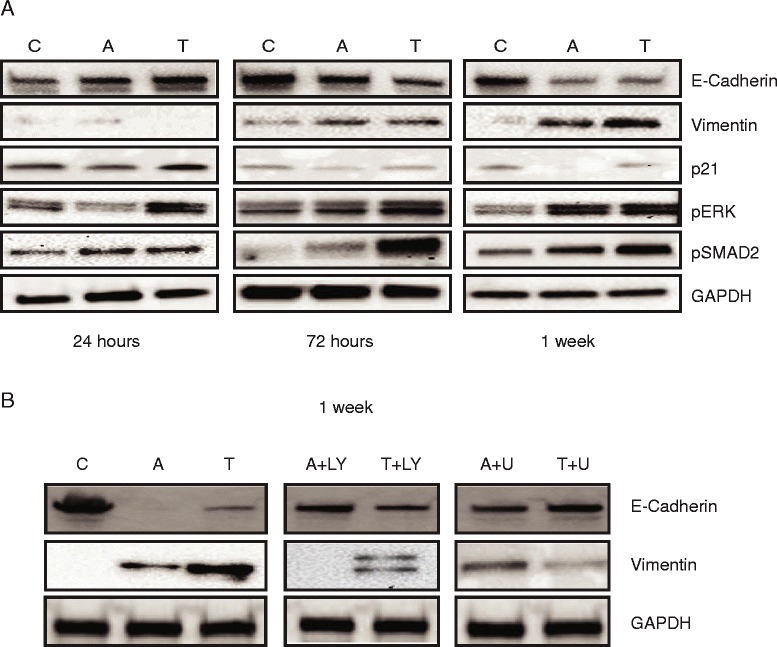
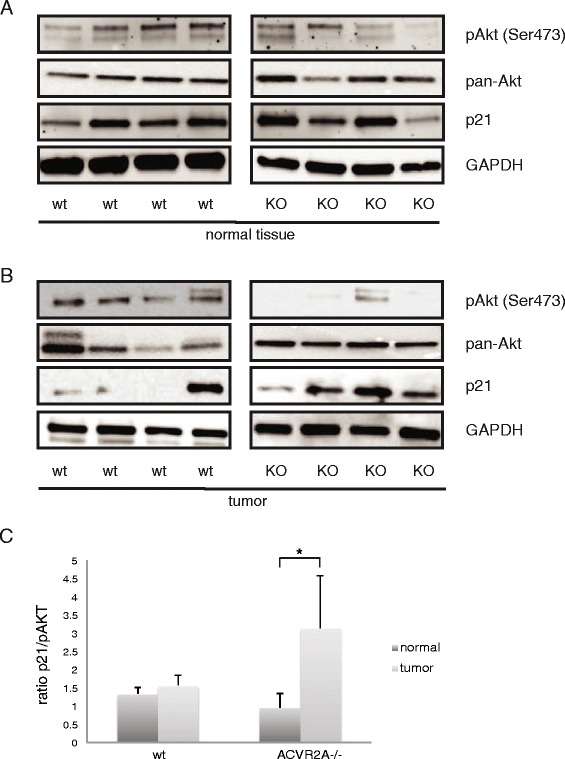
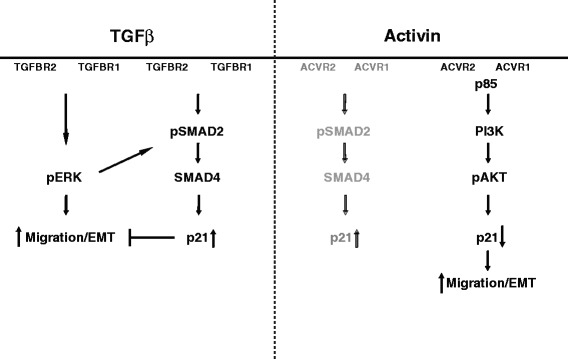
Results: In primary colon cancers, loss of nuclear p21 correlated with upstream activation of activin/PI3K while nuclear p21 expression was associated with TGFβ/MEK/ERK pathway activation. Activin, but not TGFβ, led to PI3K activation via interaction of ACVR1B and p85 independent of SMAD4, resulting in p21 downregulation. In contrast, TGFβ increased p21 via MEK/ERK pathway through a SMAD4-dependent mechanism. While activin induced EMT via PI3K, TGFβ induced EMT via MEK/ERK activation. In vivo, loss of ACVR2A resulted in loss of pAkt, consistent with activin-dependent PI3K signaling.
Conclusion: Although activin and TGFβ share growth suppressive SMAD signaling in colon cancer, they diverge in their SMAD4-independent pro-migratory signaling utilizing distinct mitogenic signaling pathways that affect EMT. p21 localization in colon cancer may determine a dominant activin versus TGFβ ligand signaling phenotype warranting further validation as a therapeutic biomarker prior to targeting TGFβ family receptors.
Figures
Similar articles
-
NFkB is essential for activin-induced colorectal cancer migration via upregulation of PI3K-MDM2 pathway.Oncotarget. 2017 Jun 6;8(23):37377-37393. doi: 10.18632/oncotarget.16343. Oncotarget. 2017. PMID: 28418896 Free PMC article.
-
Effects of activin and TGFβ on p21 in colon cancer.PLoS One. 2012;7(6):e39381. doi: 10.1371/journal.pone.0039381. Epub 2012 Jun 26. PLoS One. 2012. PMID: 22761777 Free PMC article.
-
TGFβ engages MEK/ERK to differentially regulate benign and malignant pancreas cell function.Oncogene. 2017 Jul 27;36(30):4336-4348. doi: 10.1038/onc.2016.500. Epub 2017 Apr 3. Oncogene. 2017. PMID: 28368414 Free PMC article.
-
Bone morphogenetic proteins.Growth Factors. 2004 Dec;22(4):233-41. doi: 10.1080/08977190412331279890. Growth Factors. 2004. PMID: 15621726 Review.
-
Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice.Int J Dev Biol. 2000 Apr;44(3):253-65. Int J Dev Biol. 2000. PMID: 10853822 Review.
Cited by
-
The difficulty in translating the preclinical success of combined TGFβ and immune checkpoint inhibition to clinical trial.EBioMedicine. 2022 Dec;86:104380. doi: 10.1016/j.ebiom.2022.104380. Epub 2022 Nov 28. EBioMedicine. 2022. PMID: 36455409 Free PMC article. Review.
-
Mutational profile of papillary thyroid microcarcinoma with extensive lymph node metastasis.Endocrine. 2019 Apr;64(1):130-138. doi: 10.1007/s12020-019-01842-y. Epub 2019 Jan 15. Endocrine. 2019. PMID: 30645724
-
PDCD10 promotes the tumor-supporting functions of TGF-β in pancreatic cancer.Clin Sci (Lond). 2024 Sep 18;138(18):1111-1129. doi: 10.1042/CS20240450. Clin Sci (Lond). 2024. PMID: 39212293 Free PMC article.
-
Profiling Activins and Follistatin in Colorectal Cancer According to Clinical Stage, Tumour Sidedness and Smad4 Status.Pathol Oncol Res. 2021 Nov 15;27:1610032. doi: 10.3389/pore.2021.1610032. eCollection 2021. Pathol Oncol Res. 2021. PMID: 34867090 Free PMC article.
-
Activin a promotes myofibroblast differentiation of endometrial mesenchymal stem cells via STAT3-dependent Smad/CTGF pathway.Cell Commun Signal. 2019 May 17;17(1):45. doi: 10.1186/s12964-019-0361-3. Cell Commun Signal. 2019. PMID: 31101053 Free PMC article.
References
-
- SEER Stat Fact Sheets: Colon and Rectum Cancer 2014. Available from: (http://seer.cancer.gov/statfacts/html/colorect.html).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
