

RCAN1 overexpression promotes age-dependent mitochondrial dysregulation related to neurodegeneration in Alzheimer's disease
- PMID: 26497675
- PMCID: PMC4782929
- DOI: 10.1007/s00401-015-1499-8
RCAN1 overexpression promotes age-dependent mitochondrial dysregulation related to neurodegeneration in Alzheimer's disease
Abstract
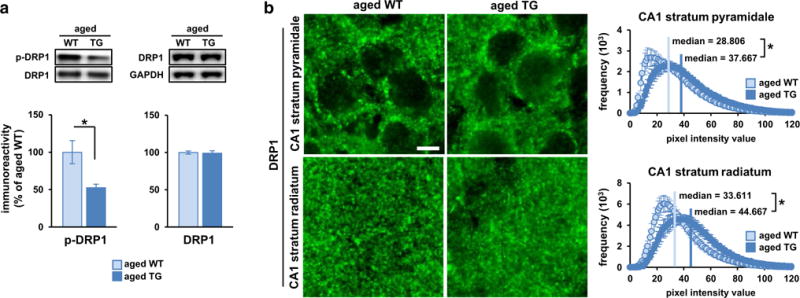
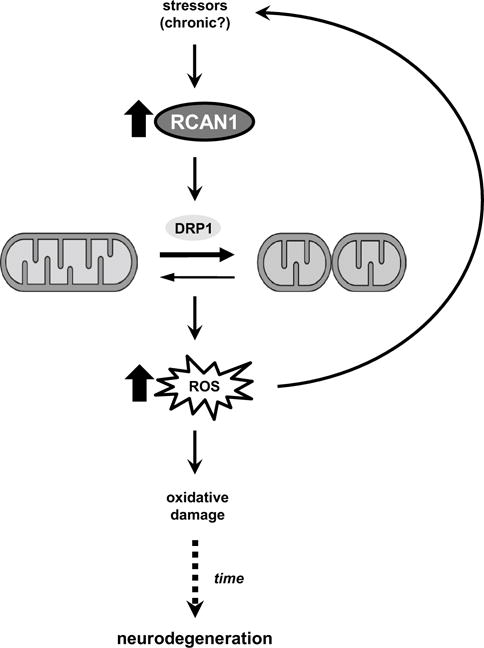
Aging is the largest risk factor for Alzheimer's disease (AD). Patients with Down syndrome (DS) develop symptoms consistent with early-onset AD, suggesting that overexpression of chromosome 21 genes such as Regulator of Calcineurin 1 (RCAN1) plays a role in AD pathogenesis. RCAN1 levels are increased in the brain of DS and AD patients but also in the human brain with normal aging. RCAN1 has been implicated in several neuronal functions, but whether its increased expression is correlative or causal in the aging-related progression of AD remains elusive. We show that brain-specific overexpression of the human RCAN1.1S isoform in mice promotes early age-dependent memory and synaptic plasticity deficits, tau pathology, and dysregulation of dynamin-related protein 1 (DRP1) activity associated with mitochondrial dysfunction and oxidative stress, reproducing key AD features. Based on these findings, we propose that chronic RCAN1 overexpression during aging alters DRP1-mediated mitochondrial fission and thus acts to promote AD-related progressive neurodegeneration.
Keywords: Aging; Calcineurin; DRP1; Fission; Mitochondria; RCAN1.
Figures







Comment in
-
Mitochondrial mRNA expression in fibroblasts of Down syndrome subjects.Hum Cell. 2018 Apr;31(2):179-181. doi: 10.1007/s13577-018-0205-2. Epub 2018 Mar 3. Hum Cell. 2018. PMID: 29502158 No abstract available.
Similar articles
-
RCAN1 knockout and overexpression recapitulate an ensemble of rest-activity and circadian disruptions characteristic of Down syndrome, Alzheimer's disease, and normative aging.J Neurodev Disord. 2022 May 24;14(1):33. doi: 10.1186/s11689-022-09444-y. J Neurodev Disord. 2022. PMID: 35610565 Free PMC article.
-
Chronic high levels of the RCAN1-1 protein may promote neurodegeneration and Alzheimer disease.Free Radic Biol Med. 2013 Sep;62:47-51. doi: 10.1016/j.freeradbiomed.2013.01.016. Epub 2013 Jan 29. Free Radic Biol Med. 2013. PMID: 23369757 Free PMC article. Review.
-
Regulator of calcineurin 1 (RCAN1) facilitates neuronal apoptosis through caspase-3 activation.J Biol Chem. 2011 Mar 18;286(11):9049-62. doi: 10.1074/jbc.M110.177519. Epub 2011 Jan 7. J Biol Chem. 2011. PMID: 21216952 Free PMC article.
-
DSCR1/RCAN1 regulates vesicle exocytosis and fusion pore kinetics: implications for Down syndrome and Alzheimer's disease.Hum Mol Genet. 2008 Apr 1;17(7):1020-30. doi: 10.1093/hmg/ddm374. Epub 2008 Jan 7. Hum Mol Genet. 2008. PMID: 18180251
-
Aberrant expression of RCAN1 in Alzheimer's pathogenesis: a new molecular mechanism and a novel drug target.Mol Neurobiol. 2014 Dec;50(3):1085-97. doi: 10.1007/s12035-014-8704-y. Epub 2014 Apr 22. Mol Neurobiol. 2014. PMID: 24752590 Review.
Cited by
-
Targeting upregulated RNA binding protein RCAN1.1: a promising strategy for neuroprotection in acute ischemic stroke.CNS Neurosci Ther. 2022 Nov;28(11):1814-1828. doi: 10.1111/cns.13921. Epub 2022 Jul 28. CNS Neurosci Ther. 2022. PMID: 35900849 Free PMC article.
-
Isoform-specific roles for AKT in affective behavior, spatial memory, and extinction related to psychiatric disorders.Elife. 2020 Dec 16;9:e56630. doi: 10.7554/eLife.56630. Elife. 2020. PMID: 33325370 Free PMC article.
-
AKT2 modulates astrocytic nicotine responses in vivo.bioRxiv [Preprint]. 2025 Jun 25:2024.05.31.596856. doi: 10.1101/2024.05.31.596856. bioRxiv. 2025. Update in: Glia. 2025 Oct;73(10):2098-2129. doi: 10.1002/glia.70059. PMID: 38854016 Free PMC article. Updated. Preprint.
-
Overexpression of RCAN1, a Gene on Human Chromosome 21, Alters Cell Redox and Mitochondrial Function in Enamel Cells.Cells. 2022 Nov 11;11(22):3576. doi: 10.3390/cells11223576. Cells. 2022. PMID: 36429004 Free PMC article.
-
RCAN1 Knockdown Reverts Defects in the Number of Calcium-Induced Exocytotic Events in a Cellular Model of Down Syndrome.Front Cell Neurosci. 2018 Jul 6;12:189. doi: 10.3389/fncel.2018.00189. eCollection 2018. Front Cell Neurosci. 2018. PMID: 30034324 Free PMC article.
References
-
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
