

High-throughput genotyping of CRISPR/Cas9-mediated mutants using fluorescent PCR-capillary gel electrophoresis
- PMID: 26498861
- PMCID: PMC4620477
- DOI: 10.1038/srep15587
High-throughput genotyping of CRISPR/Cas9-mediated mutants using fluorescent PCR-capillary gel electrophoresis
Abstract
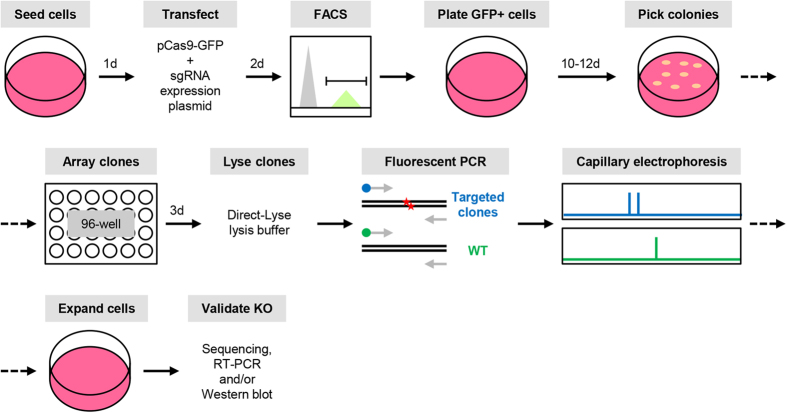
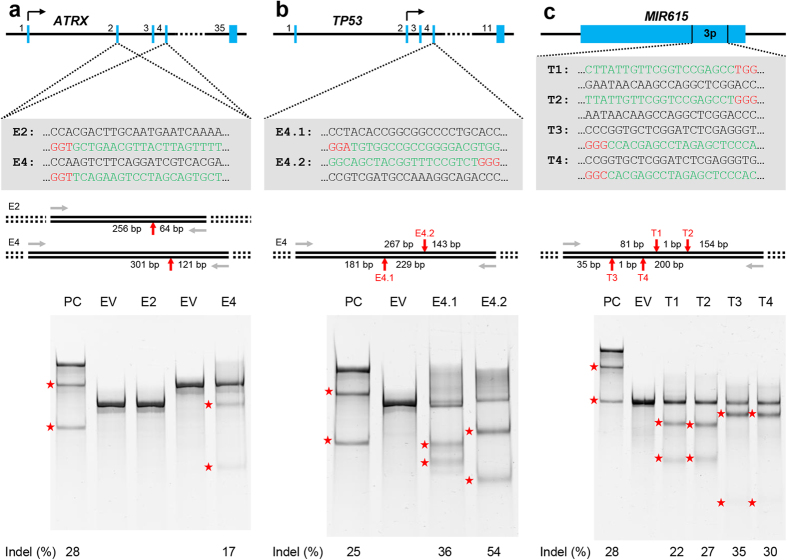
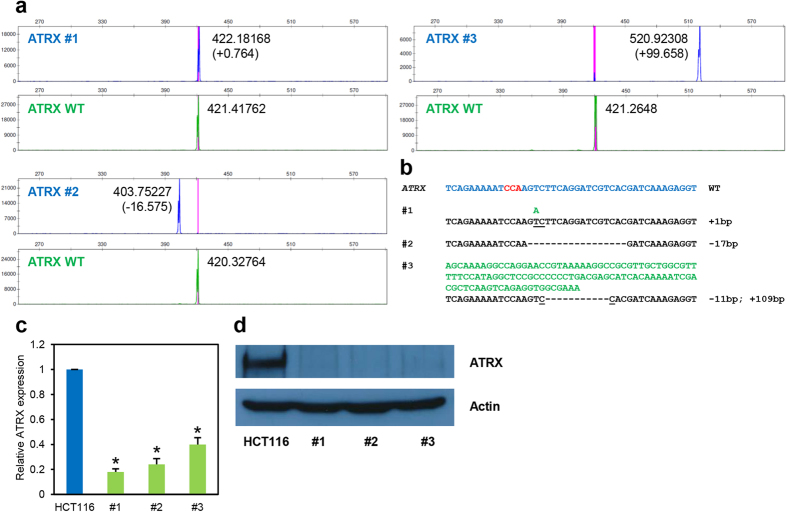
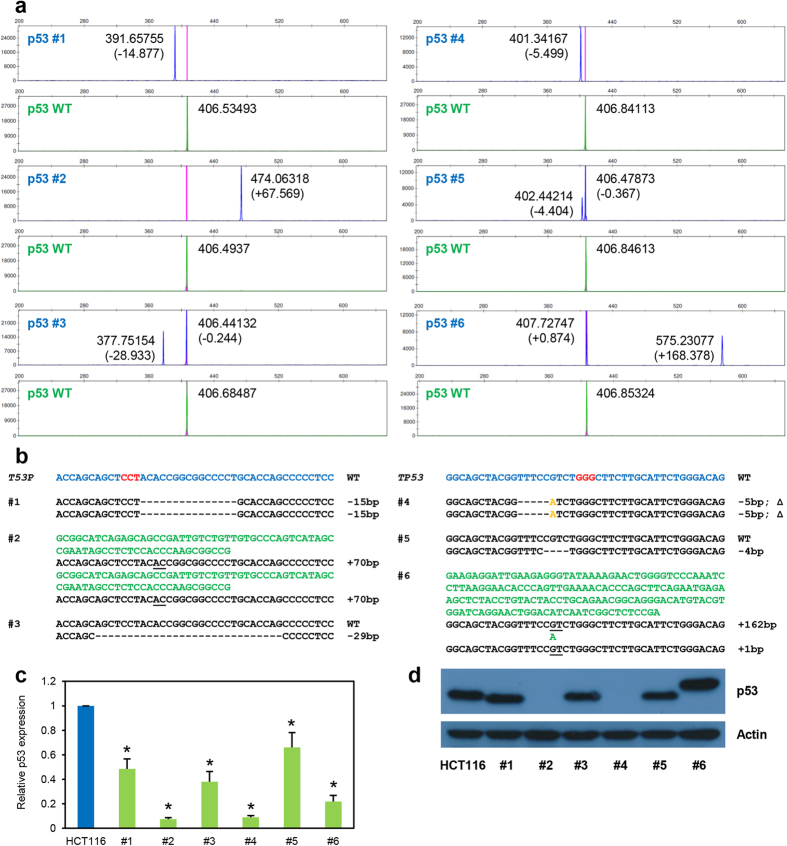
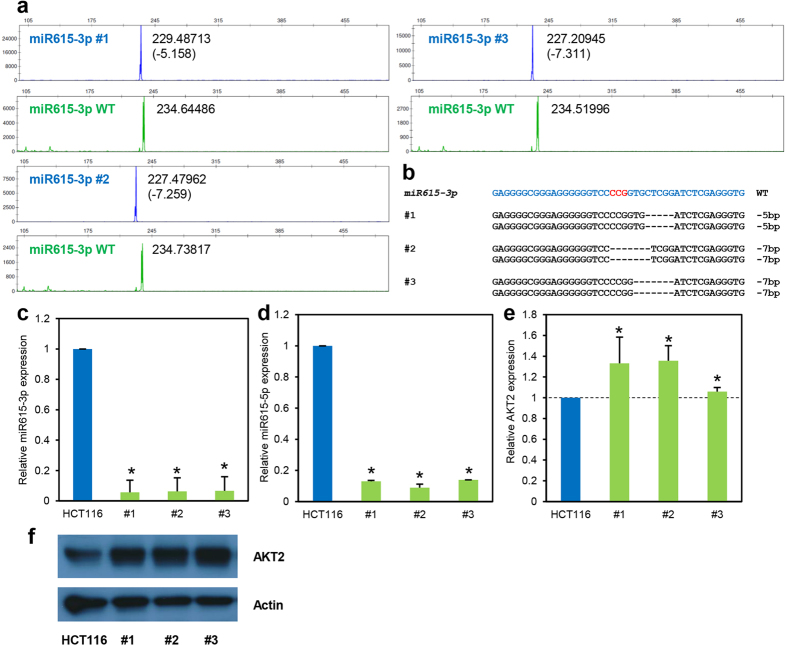
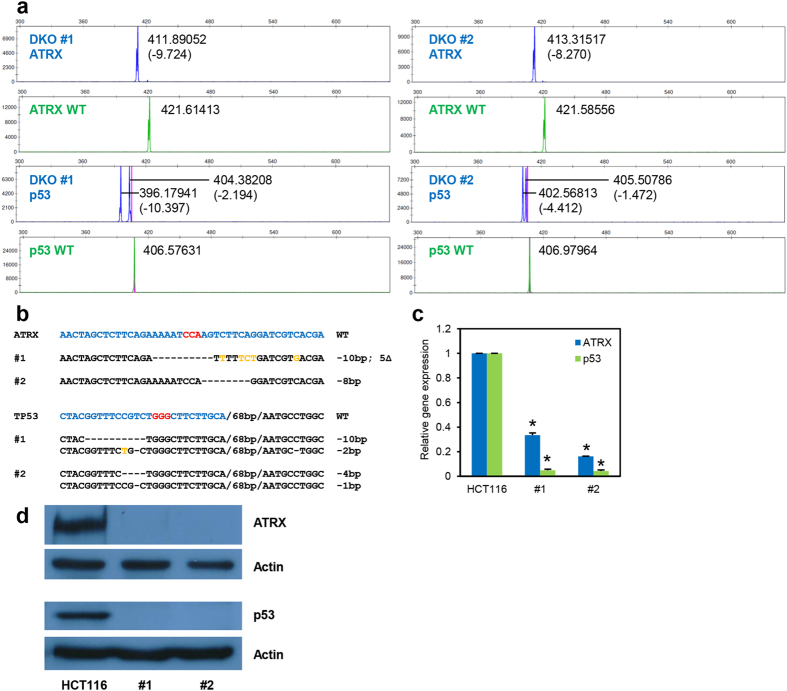
Recent advances in the engineering of sequence-specific synthetic nucleases provide enormous opportunities for genetic manipulation of gene expression in order to study their cellular function in vivo. However, current genotyping methods to detect these programmable nuclease-induced insertion/deletion (indel) mutations in targeted human cells are not compatible for high-throughput screening of knockout clones due to inherent limitations and high cost. Here, we describe an efficient method of genotyping clonal CRISPR/Cas9-mediated mutants in a high-throughput manner involving the use of a direct lysis buffer to extract crude genomic DNA straight from cells in culture, and fluorescent PCR coupled with capillary gel electrophoresis. This technique also allows for genotyping of multiplexed gene targeting in a single clone. Overall, this time- and cost-saving technique is able to circumvent the limitations of current genotyping methods and support high-throughput screening of nuclease-induced mutants.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
