

Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages
- PMID: 26502406
- PMCID: PMC4653072
- DOI: 10.1038/ni.3299
Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages
Abstract
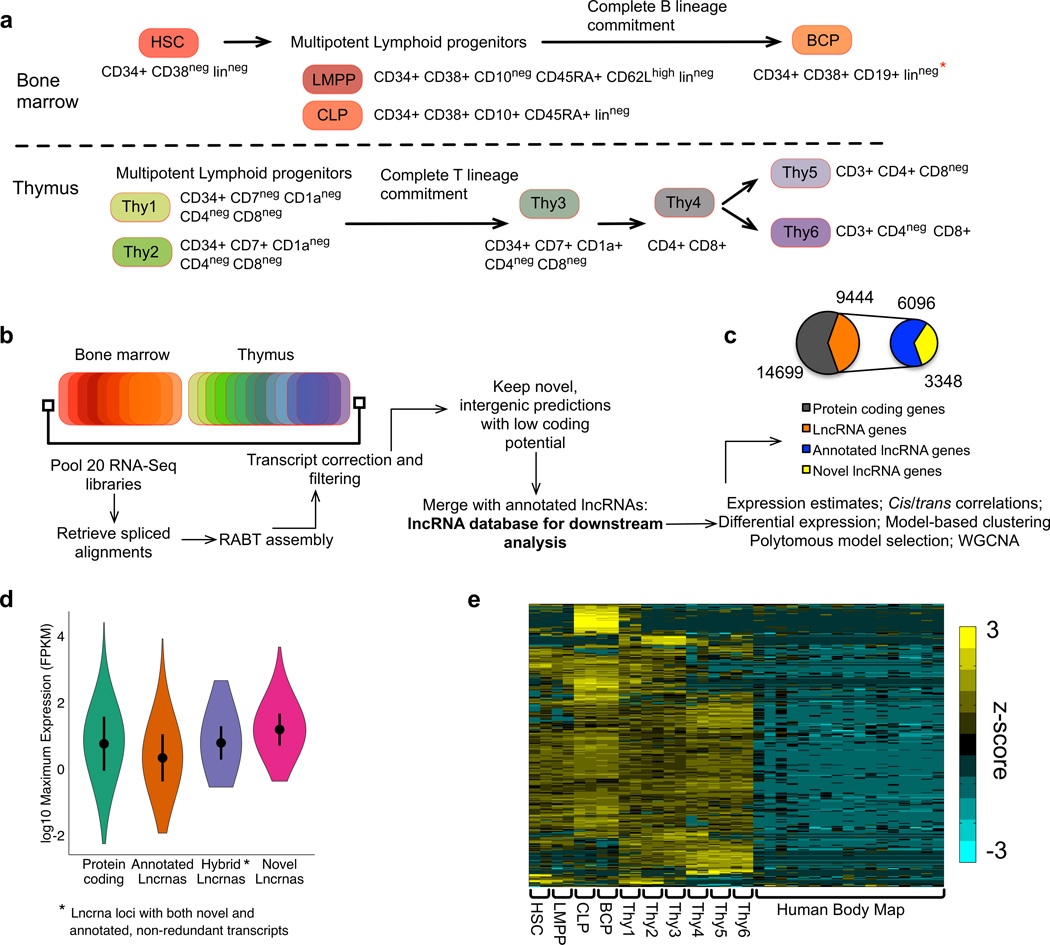
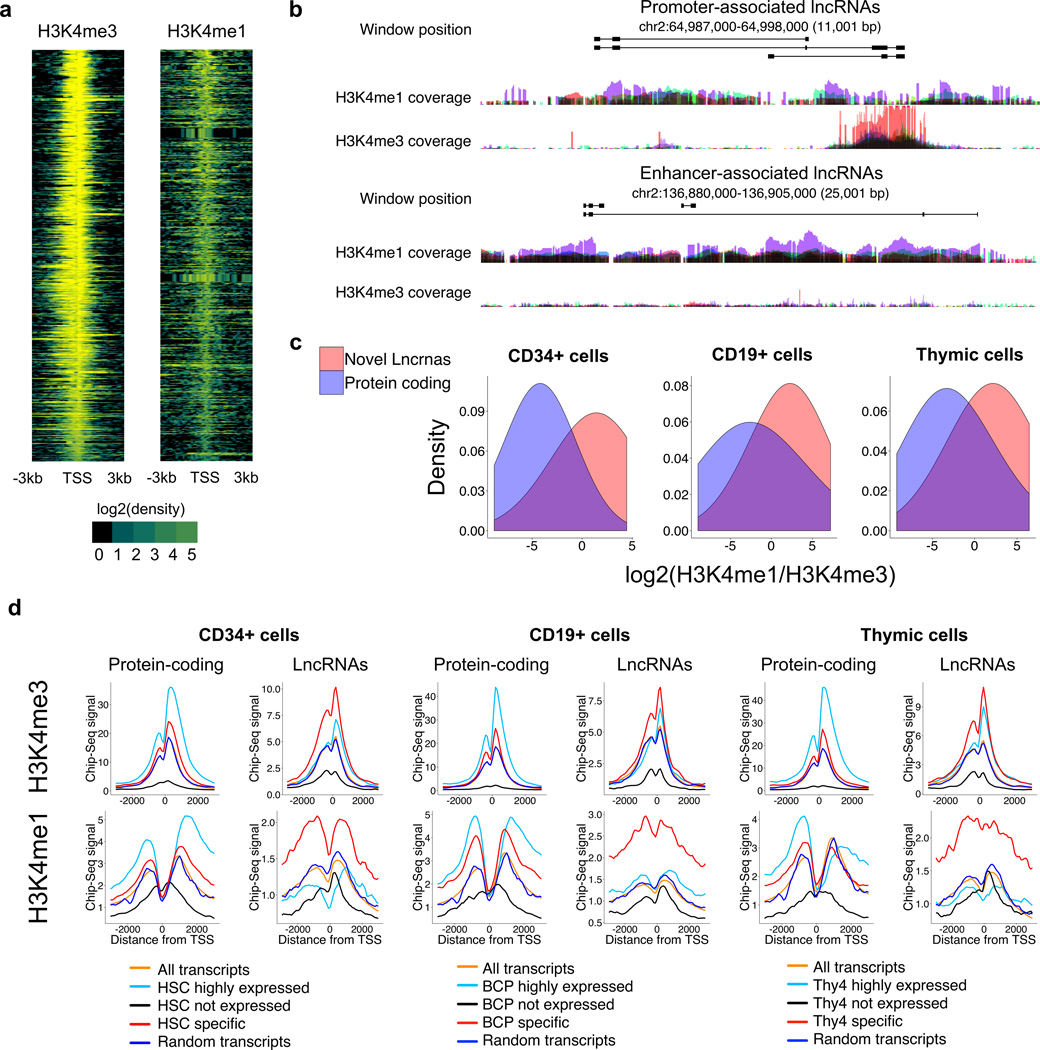
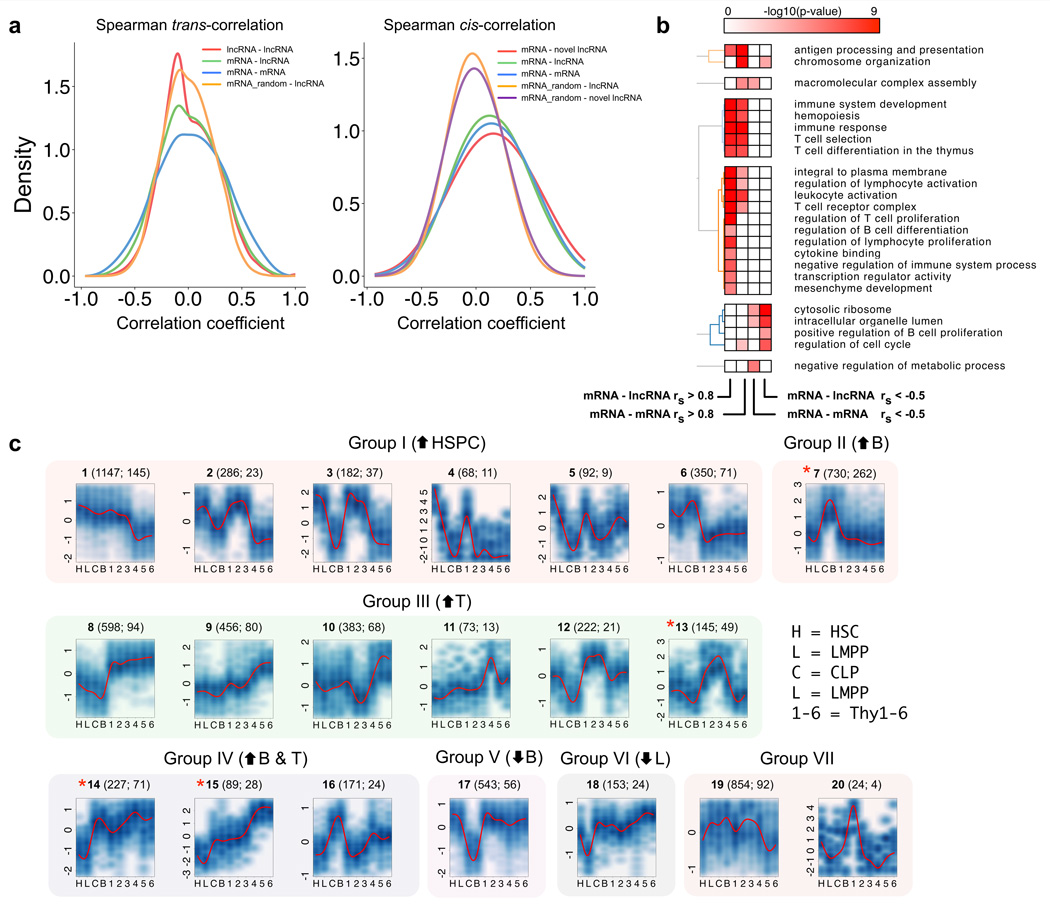
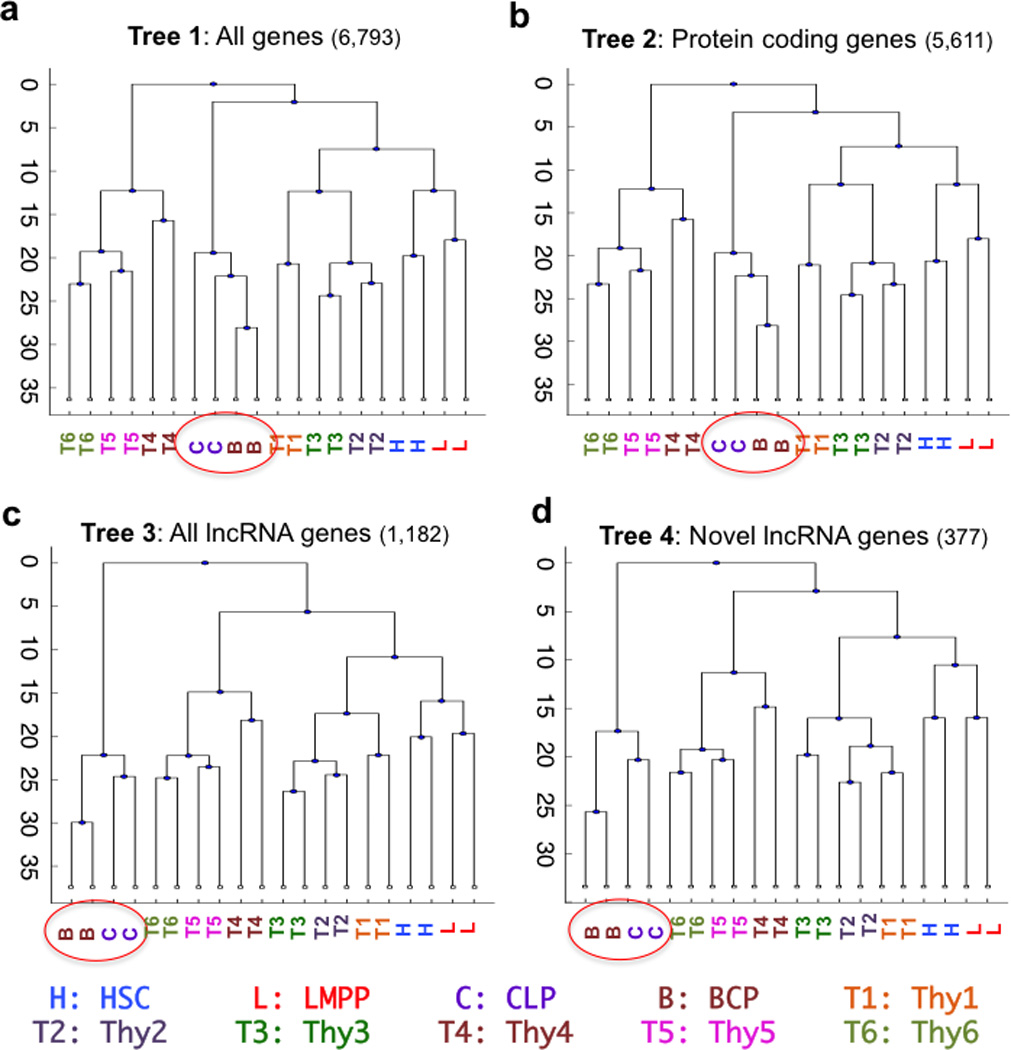
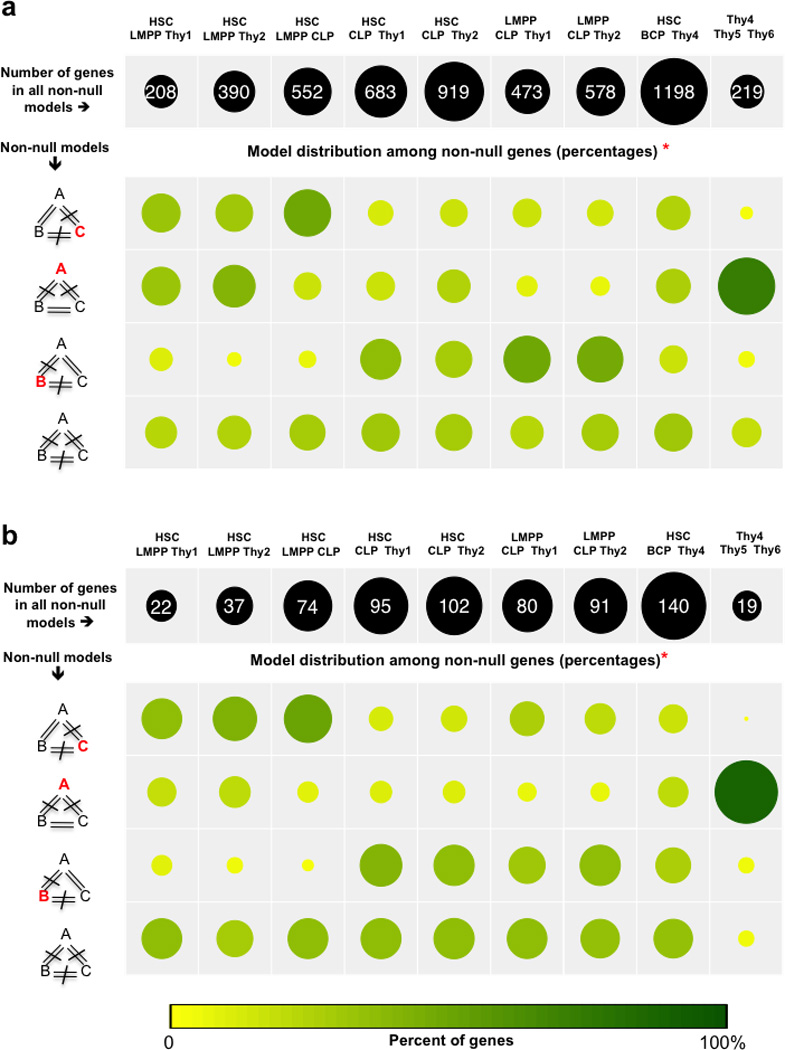
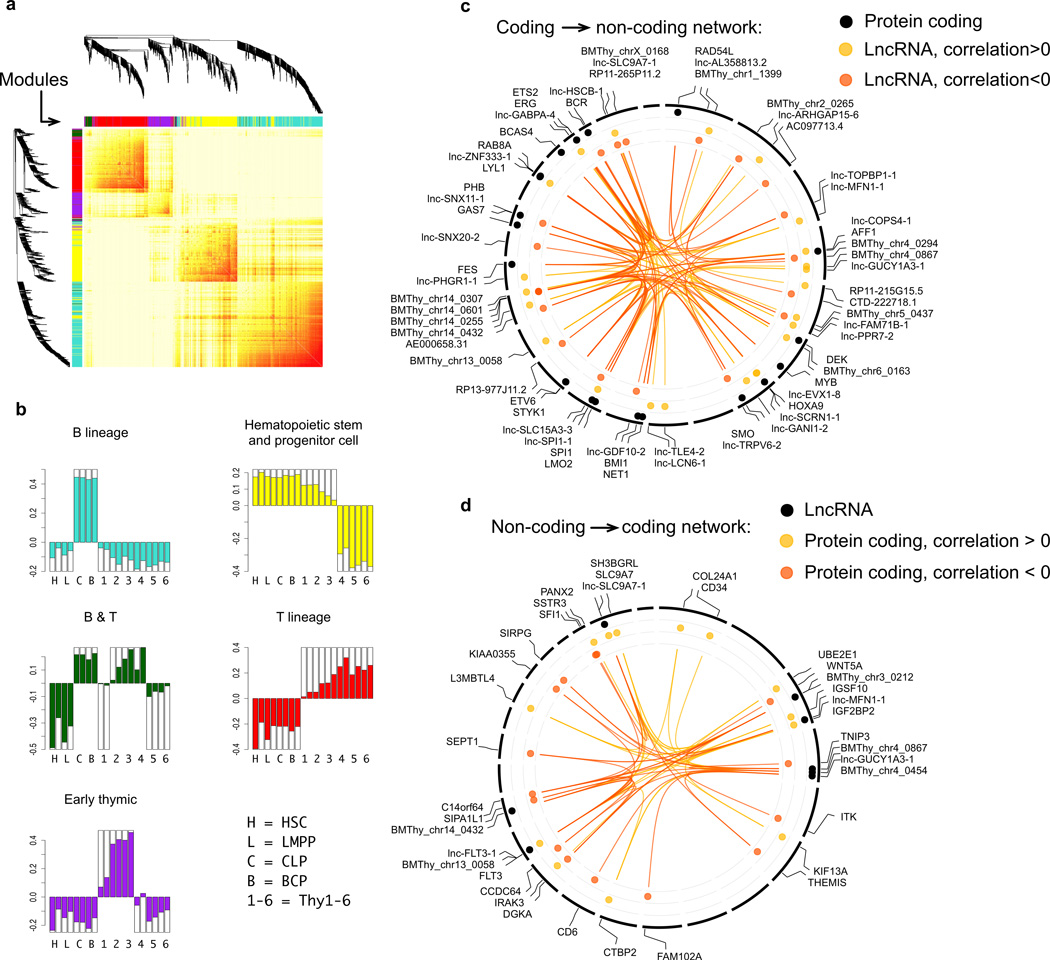
To elucidate the transcriptional 'landscape' that regulates human lymphoid commitment during postnatal life, we used RNA sequencing to assemble the long non-coding transcriptome across human bone marrow and thymic progenitor cells spanning the earliest stages of B lymphoid and T lymphoid specification. Over 3,000 genes encoding previously unknown long non-coding RNAs (lncRNAs) were revealed through the analysis of these rare populations. Lymphoid commitment was characterized by lncRNA expression patterns that were highly stage specific and were more lineage specific than those of protein-coding genes. Protein-coding genes co-expressed with neighboring lncRNA genes showed enrichment for ontologies related to lymphoid differentiation. The exquisite cell-type specificity of global lncRNA expression patterns independently revealed new developmental relationships among the earliest progenitor cells in the human bone marrow and thymus.
Figures
References
-
- Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. - PubMed
-
- Galy A, Travis M, Cen D, Chen B, Human TB. natural killer, and dendritic cells arise from a common bone marrow progenitor cell subset. Immunity. 1995;3:459–473. - PubMed
-
- Plum J, et al. Human intrathymic development: a selective approach. Semin Immunopathol. 2008;30:411–423. - PubMed
-
- Doulatov S, et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11:585–593. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
