

Integrated Bottom-Up and Top-Down Proteomics of Patient-Derived Breast Tumor Xenografts
- PMID: 26503891
- PMCID: PMC4762530
- DOI: 10.1074/mcp.M114.047480
Integrated Bottom-Up and Top-Down Proteomics of Patient-Derived Breast Tumor Xenografts
Abstract
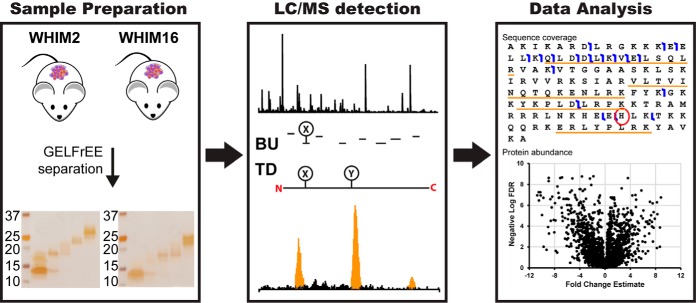
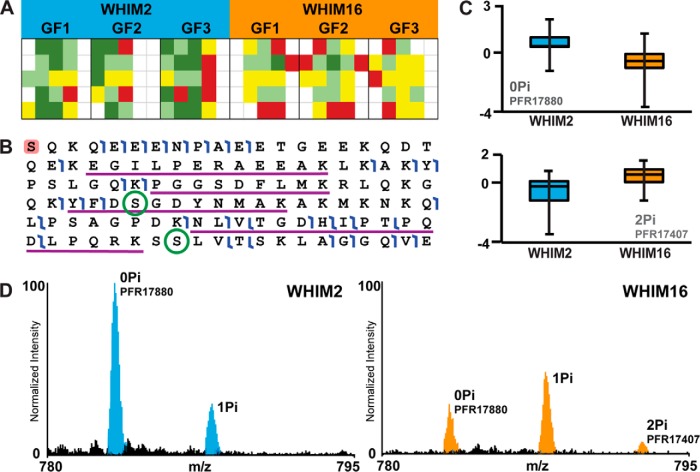
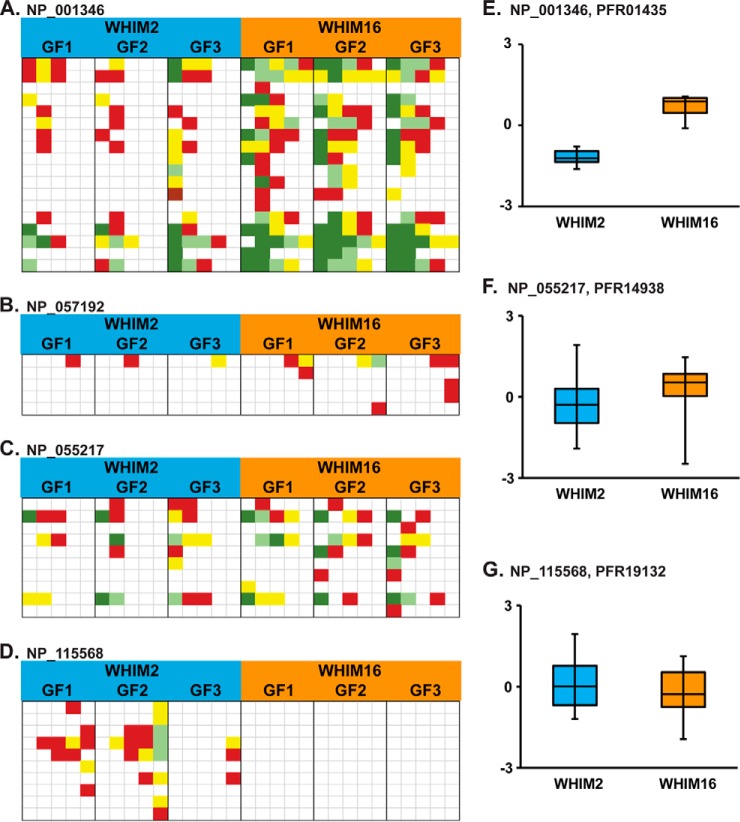
Bottom-up proteomics relies on the use of proteases and is the method of choice for identifying thousands of protein groups in complex samples. Top-down proteomics has been shown to be robust for direct analysis of small proteins and offers a solution to the "peptide-to-protein" inference problem inherent with bottom-up approaches. Here, we describe the first large-scale integration of genomic, bottom-up and top-down proteomic data for the comparative analysis of patient-derived mouse xenograft models of basal and luminal B human breast cancer, WHIM2 and WHIM16, respectively. Using these well-characterized xenograft models established by the National Cancer Institute's Clinical Proteomic Tumor Analysis Consortium, we compared and contrasted the performance of bottom-up and top-down proteomics to detect cancer-specific aberrations at the peptide and proteoform levels and to measure differential expression of proteins and proteoforms. Bottom-up proteomic analysis of the tumor xenografts detected almost 10 times as many coding nucleotide polymorphisms and peptides resulting from novel splice junctions than top-down. For proteins in the range of 0-30 kDa, where quantitation was performed using both approaches, bottom-up proteomics quantified 3,519 protein groups from 49,185 peptides, while top-down proteomics quantified 982 proteoforms mapping to 358 proteins. Examples of both concordant and discordant quantitation were found in a ∼60:40 ratio, providing a unique opportunity for top-down to fill in missing information. The two techniques showed complementary performance, with bottom-up yielding eight times more identifications of 0-30 kDa proteins in xenograft proteomes, but failing to detect differences in certain posttranslational modifications (PTMs), such as phosphorylation pattern changes of alpha-endosulfine. This work illustrates the potency of a combined bottom-up and top-down proteomics approach to deepen our knowledge of cancer biology, especially when genomic data are available.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures


References
-
- Balmain A., Gray J., and Ponder B. (2003) The genetics and genomics of cancer. Nat. Genet. 33, 238–244 - PubMed
-
- Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M. C., Zimmerman L. J., Shaddox K. F., Kim S., Davies S. R., Wang S., Wang P., Kinsinger C. R., Rivers R. C., Rodriguez H., Townsend R. R., Ellis M. J., Carr S. A., Tabb D. L., Coffey R. J., Slebos R. J., and Liebler D. C. (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387 - PMC - PubMed
-
- Zhang Z., Wu S., Stenoien D. L., and Pasa-Tolic L. (2014) High-throughput proteomics. Annu. Rev. Anal. Chem. 7, 427–454 - PubMed
-
- Wiśniewski J. R., Zougman A., and Mann M. (2009) Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 8, 5674–5678 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
