

Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy
- PMID: 26503985
- PMCID: PMC4648202
- DOI: 10.1093/cvr/cvv238
Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy
Abstract
Aims: Nicotinamide adenine dinucleotide oxidases (NOXs) are important contributors to cellular oxidative stress in the cardiovascular system. The NOX2 isoform is upregulated in numerous disorders, including dystrophic cardiomyopathy, where it drives the progression of the disease. However, mechanisms underlying NOX2 overexpression are still unknown. We investigated the role of microRNAs (miRs) in the regulation of NOX2 expression.
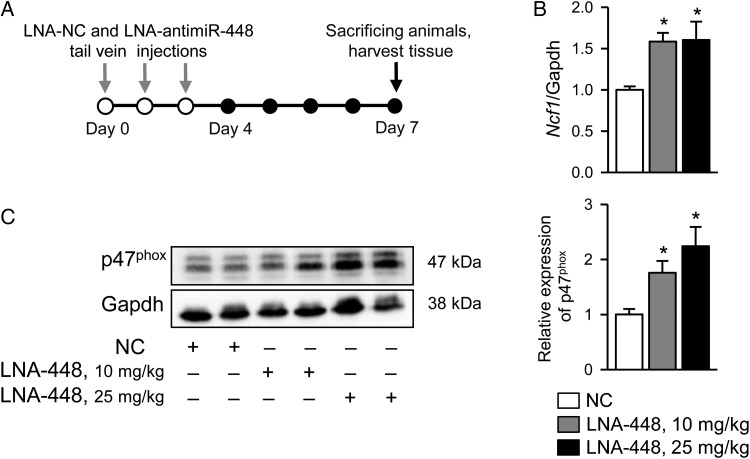
Methods and results: Duchenne muscular dystrophy (DMD) was used as a model of cardiomyopathy. After screening with miRNA target prediction databases and following qRT-PCR analysis, we found drastic downregulation of miR-448-3p in hearts of mdx mice, an animal model of DMD. The downregulation correlated with overexpression of the Ncf1 gene, encoding the NOX2 regulatory subunit p47(phox). Specificity of Ncf1 targeting by miR-448-3p was validated by luciferase reporter assay. Silencing of miR-448-3p in wild-type mice had a dramatic effect on cellular and functional properties of cardiac muscle as assessed by western blotting, qRT-PCR, confocal imaging, echocardiography, and histology. Acute treatment of mice with LNA-miR-448 inhibitors led to increased Ncf1 expression, abnormally elevated reactive oxygen species (ROS) production and exacerbated Ca(2+) signalling in cardiomyocytes, reminiscent of features previously observed in dystrophic cardiac cells. In addition, chronic inhibition of miR-448-3p resulted in dilated cardiomyopathy and arrhythmia, hallmarks of dystrophic cardiomyopathy.
Conclusions: Our studies suggest that downregulation of miR-448-3p leads to the increase in the expression of Ncf1 gene and p47(phox) protein, as well as to the substantial increase in NOX2-derived ROS production. Cellular oxidative stress subsequently triggers events that finally culminate in cardiac tissue damage and development of cardiomyopathy.
Keywords: Cardiomyopathy; Dystrophin; NOX; Oxidative stress; microRNA.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author 2015. For permissions please email: journals.permissions@oup.com.
Figures





References
-
- Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care 2008;31:S170–S180. - PubMed
-
- Nabeebaccus A, Zhang M, Shah AM. NADPH oxidases and cardiac remodelling. Heart Fail Rev 2010;16:5–12. - PubMed
-
- Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245–313. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
