

Longitudinal Antigenic Sequences and Sites from Intra-Host Evolution (LASSIE) Identifies Immune-Selected HIV Variants
- PMID: 26506369
- PMCID: PMC4632389
- DOI: 10.3390/v7102881
Longitudinal Antigenic Sequences and Sites from Intra-Host Evolution (LASSIE) Identifies Immune-Selected HIV Variants
Abstract
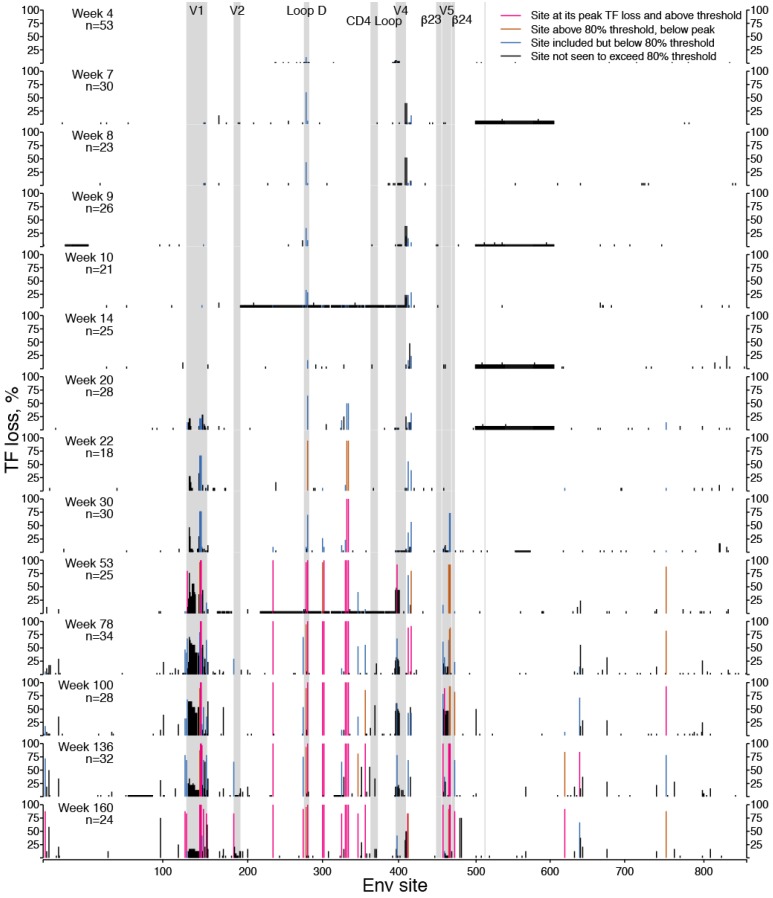
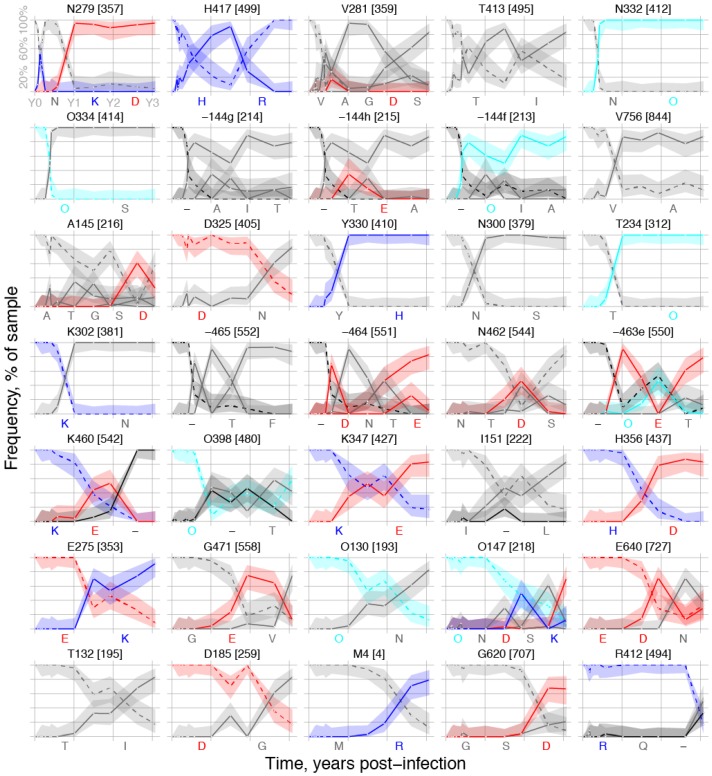
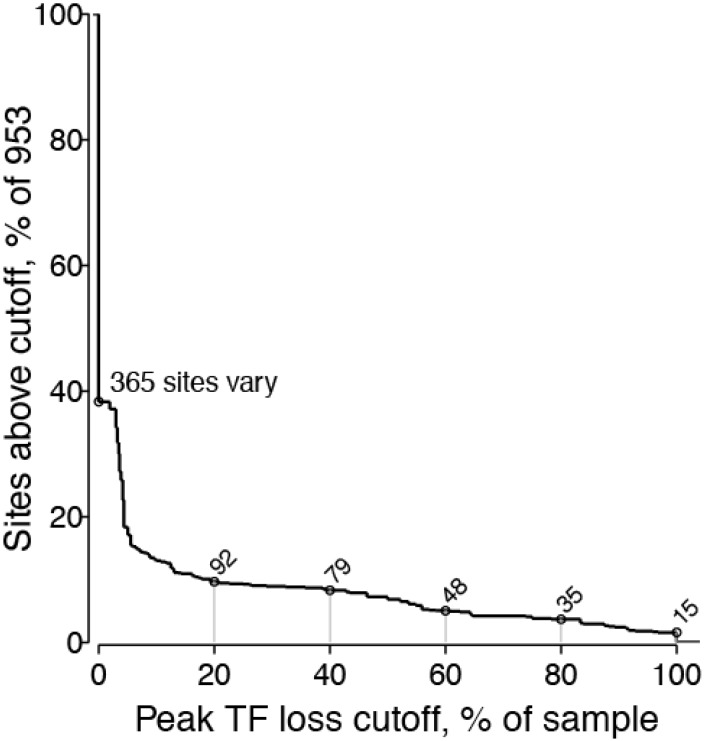
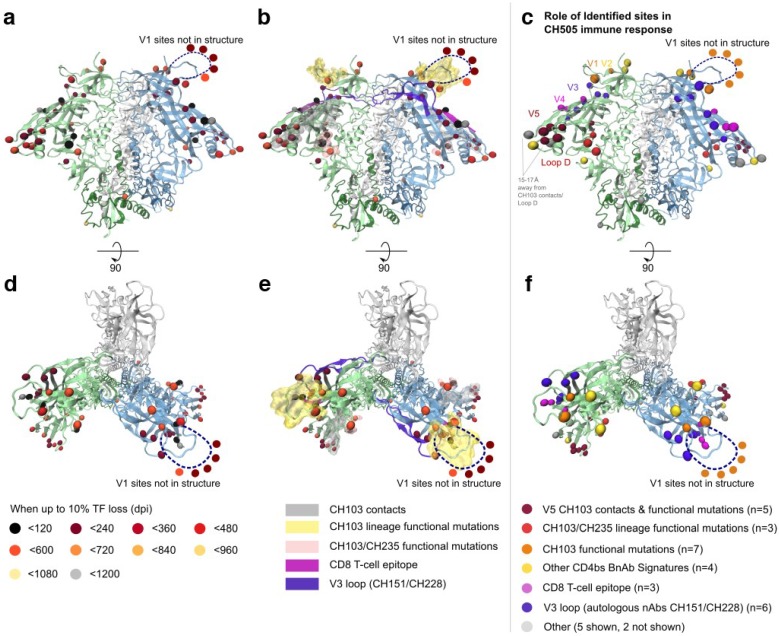
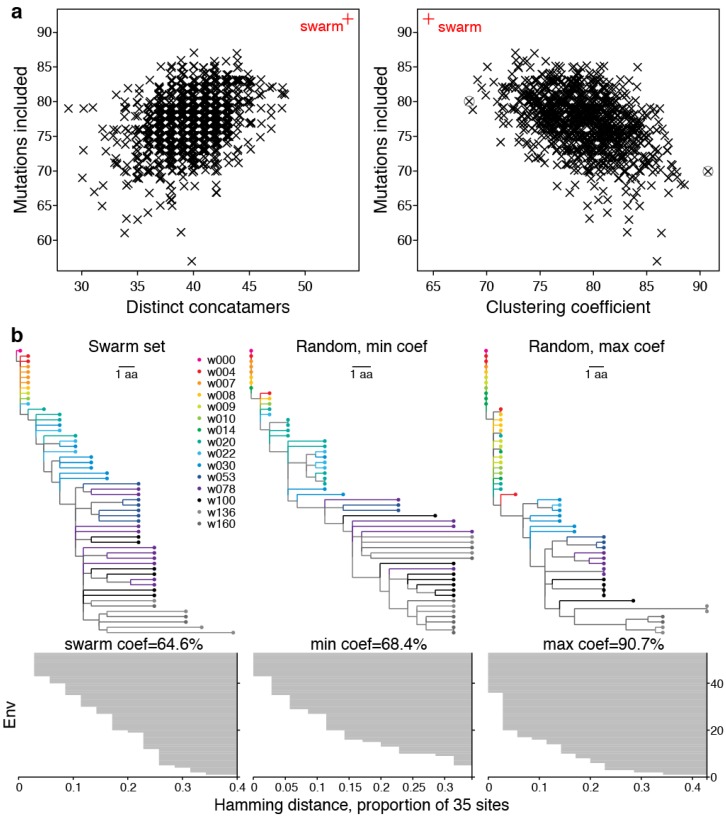
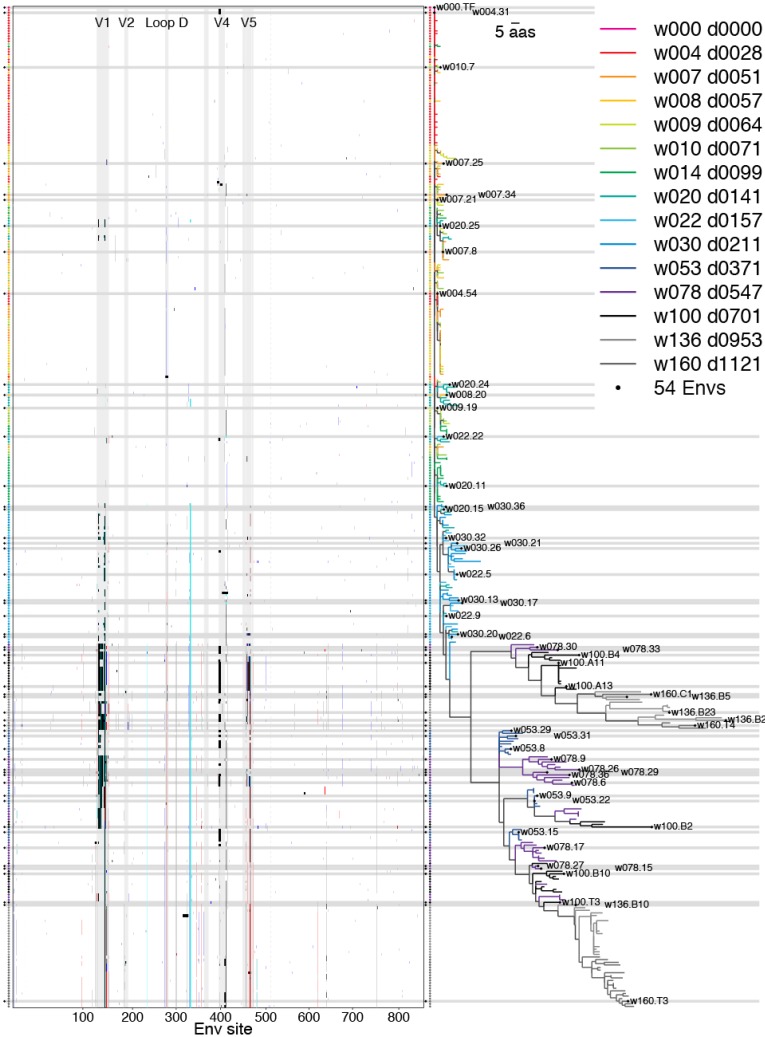
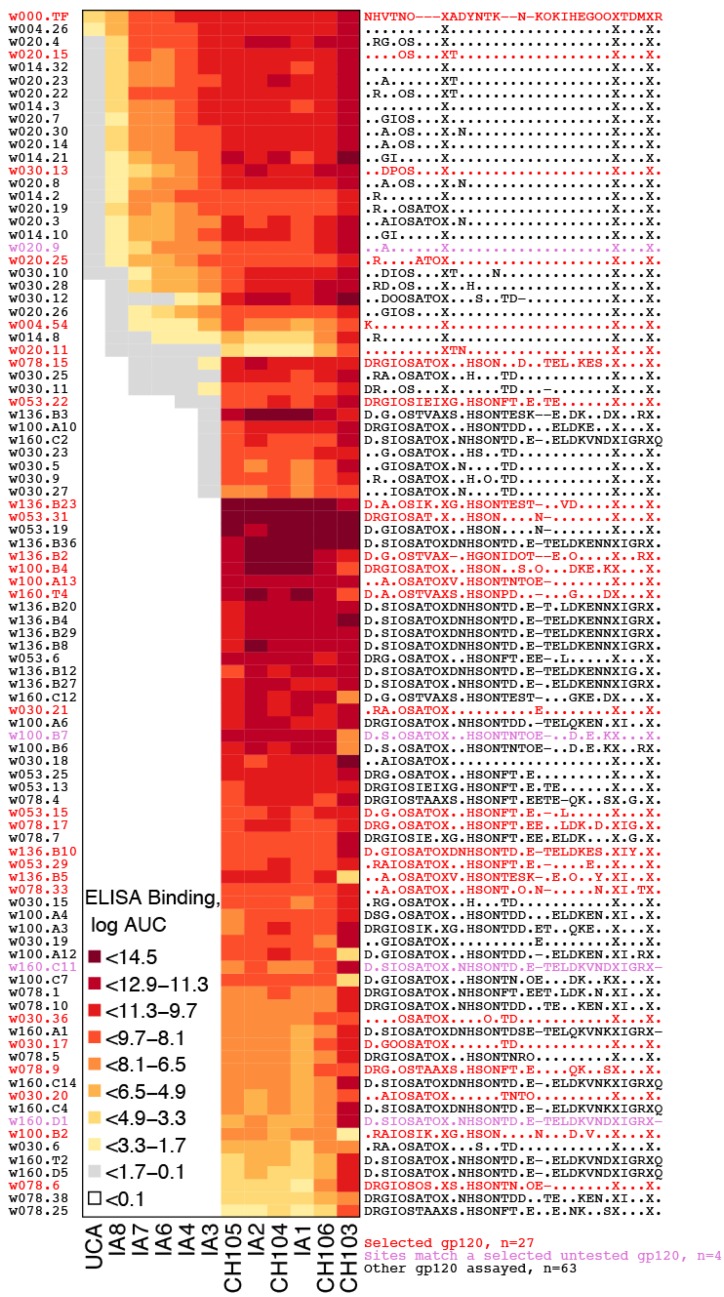
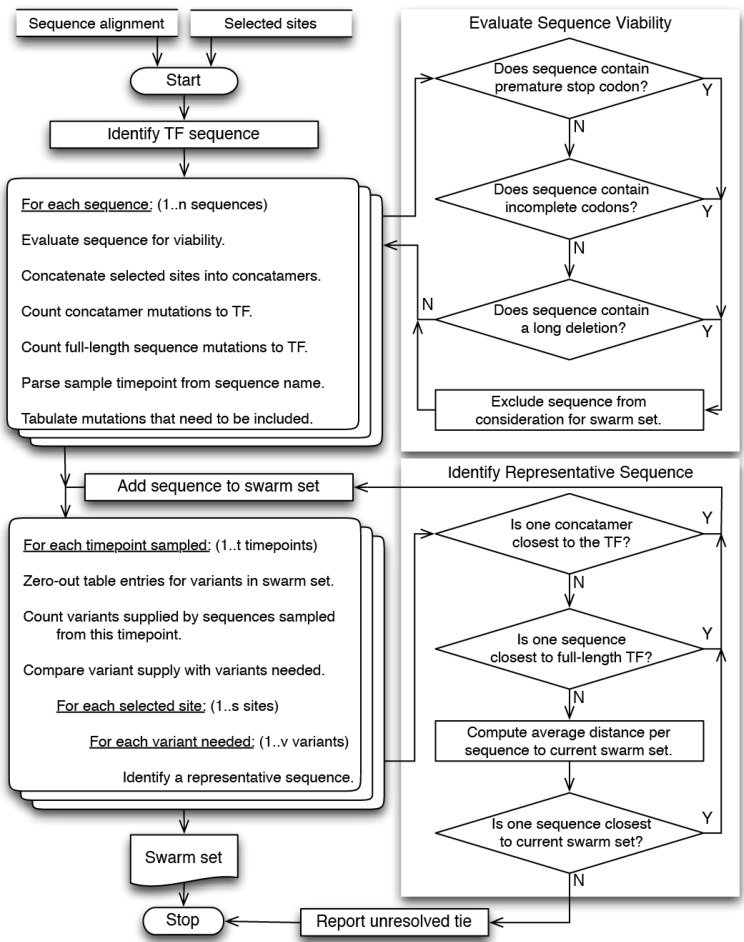
Within-host genetic sequencing from samples collected over time provides a dynamic view of how viruses evade host immunity. Immune-driven mutations might stimulate neutralization breadth by selecting antibodies adapted to cycles of immune escape that generate within-subject epitope diversity. Comprehensive identification of immune-escape mutations is experimentally and computationally challenging. With current technology, many more viral sequences can readily be obtained than can be tested for binding and neutralization, making down-selection necessary. Typically, this is done manually, by picking variants that represent different time-points and branches on a phylogenetic tree. Such strategies are likely to miss many relevant mutations and combinations of mutations, and to be redundant for other mutations. Longitudinal Antigenic Sequences and Sites from Intrahost Evolution (LASSIE) uses transmitted founder loss to identify virus "hot-spots" under putative immune selection and chooses sequences that represent recurrent mutations in selected sites. LASSIE favors earliest sequences in which mutations arise. With well-characterized longitudinal Env sequences, we confirmed selected sites were concentrated in antibody contacts and selected sequences represented diverse antigenic phenotypes. Practical applications include rapidly identifying immune targets under selective pressure within a subject, selecting minimal sets of reagents for immunological assays that characterize evolving antibody responses, and for immunogens in polyvalent "cocktail" vaccines.
Keywords: antigenic swarm; coevolution; envelope glycoprotein; human immunodeficiency virus type 1; immune escape; immunogen design; neutralizing antibodies; quasispecies; selection; vaccine.
Figures

References
-
- Mascola J.R., Lewis M.G., Stiegler G., Harris D., VanCott T.C., Hayes D., Louder M.K., Brown C.R., Sapan C.V., Frankel S.S., et al. Protection of macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J. Virol. 1999;73:4009–4018. - PMC - PubMed
-
- Moldt B., Rakasz E.G., Schultz N., Chan-Hui P.Y., Swiderek K., Weisgrau K.L., Piaskowski S.M., Bergman Z., Watkins D.I., Poignard P., et al. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc. Natl. Acad. Sci. USA. 2012;109:18921–18925. - PMC - PubMed
-
- Keele B., Giorgi E., Salazar-Gonzalez J., Decker J., Pham K., Salazar M., Sun C., Grayson T., Wang S., Li H., et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
