

Predicting the Effect of Mutations on Protein-Protein Binding Interactions through Structure-Based Interface Profiles
- PMID: 26506533
- PMCID: PMC4624718
- DOI: 10.1371/journal.pcbi.1004494
Predicting the Effect of Mutations on Protein-Protein Binding Interactions through Structure-Based Interface Profiles
Abstract
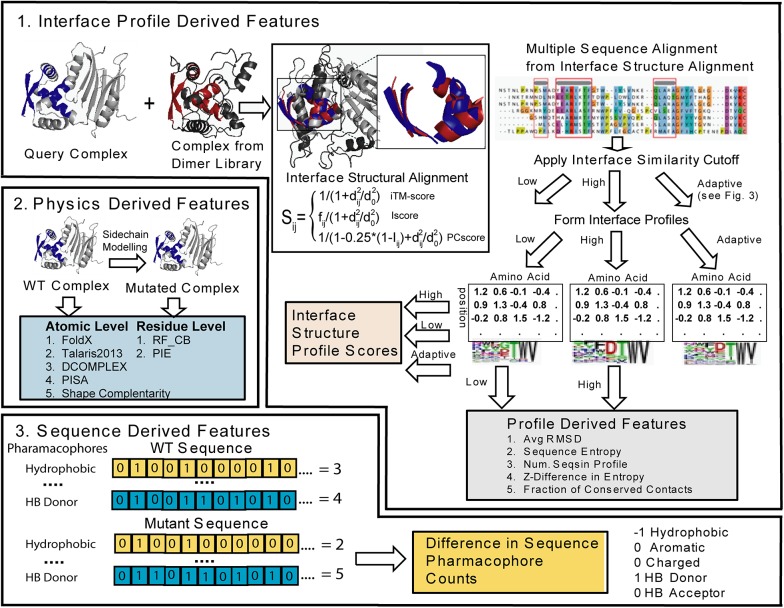
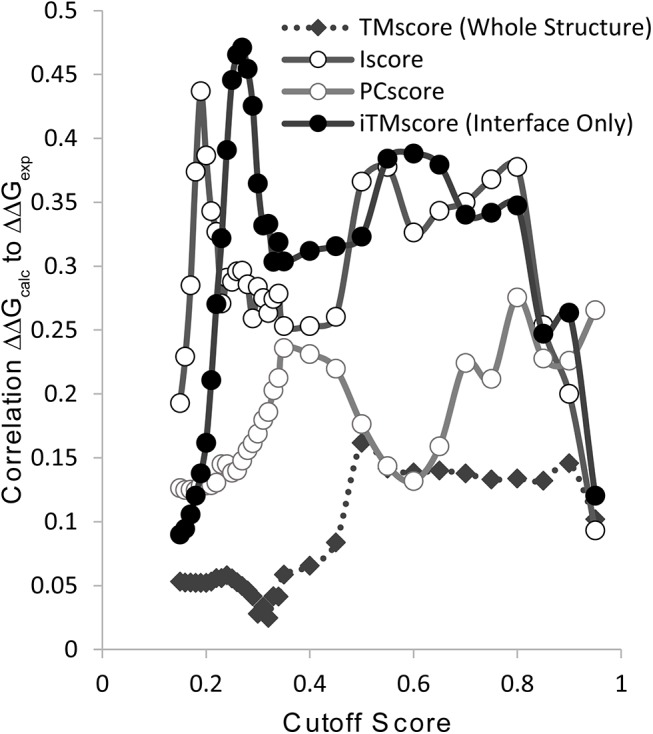
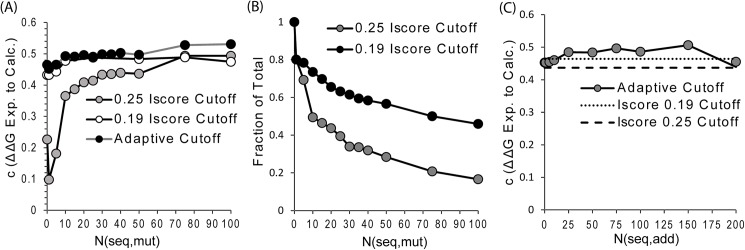
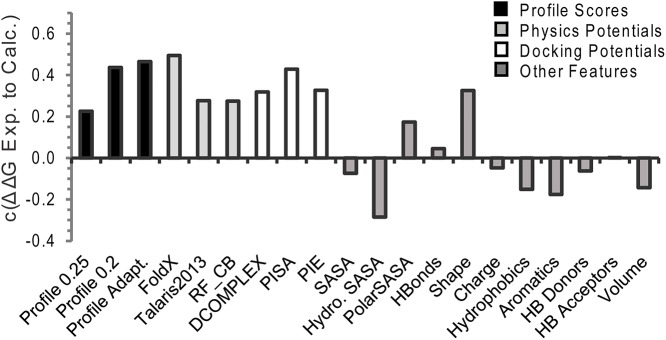
The formation of protein-protein complexes is essential for proteins to perform their physiological functions in the cell. Mutations that prevent the proper formation of the correct complexes can have serious consequences for the associated cellular processes. Since experimental determination of protein-protein binding affinity remains difficult when performed on a large scale, computational methods for predicting the consequences of mutations on binding affinity are highly desirable. We show that a scoring function based on interface structure profiles collected from analogous protein-protein interactions in the PDB is a powerful predictor of protein binding affinity changes upon mutation. As a standalone feature, the differences between the interface profile score of the mutant and wild-type proteins has an accuracy equivalent to the best all-atom potentials, despite being two orders of magnitude faster once the profile has been constructed. Due to its unique sensitivity in collecting the evolutionary profiles of analogous binding interactions and the high speed of calculation, the interface profile score has additional advantages as a complementary feature to combine with physics-based potentials for improving the accuracy of composite scoring approaches. By incorporating the sequence-derived and residue-level coarse-grained potentials with the interface structure profile score, a composite model was constructed through the random forest training, which generates a Pearson correlation coefficient >0.8 between the predicted and observed binding free-energy changes upon mutation. This accuracy is comparable to, or outperforms in most cases, the current best methods, but does not require high-resolution full-atomic models of the mutant structures. The binding interface profiling approach should find useful application in human-disease mutation recognition and protein interface design studies.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
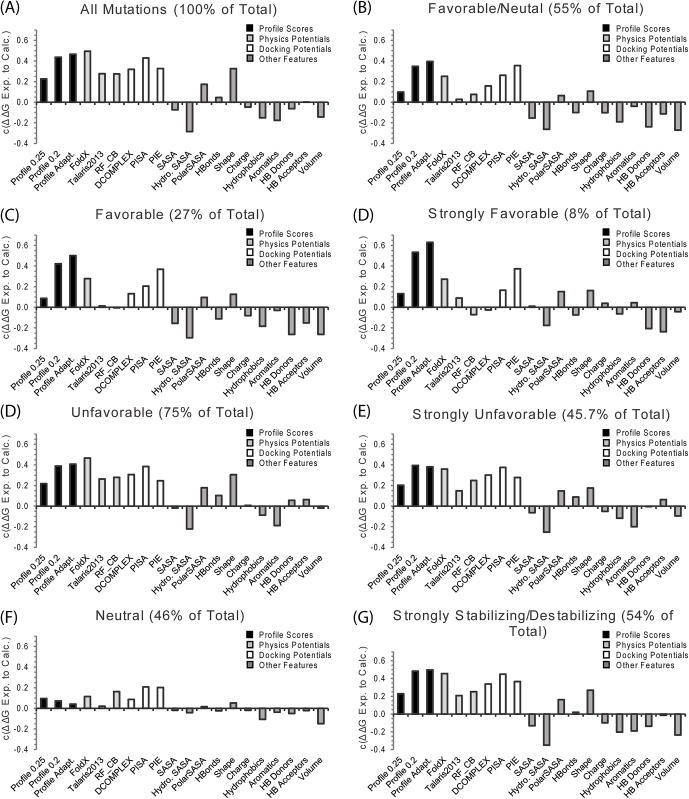
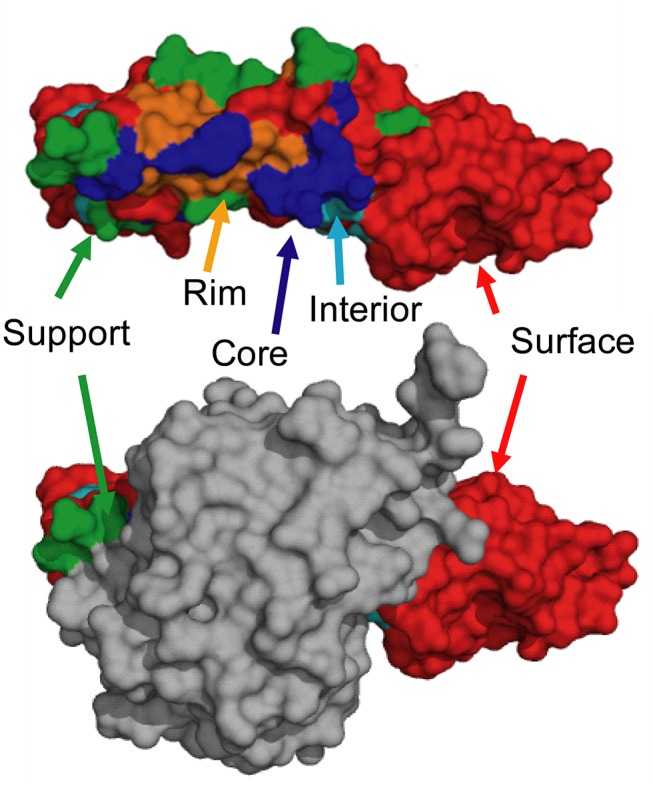
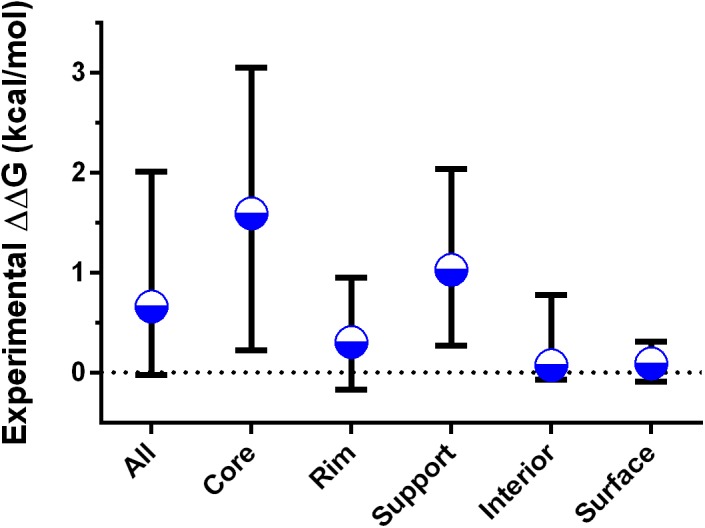
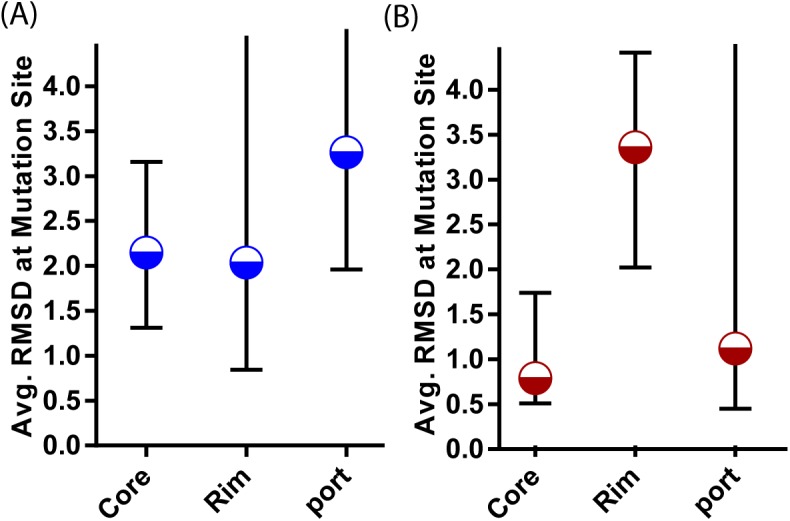
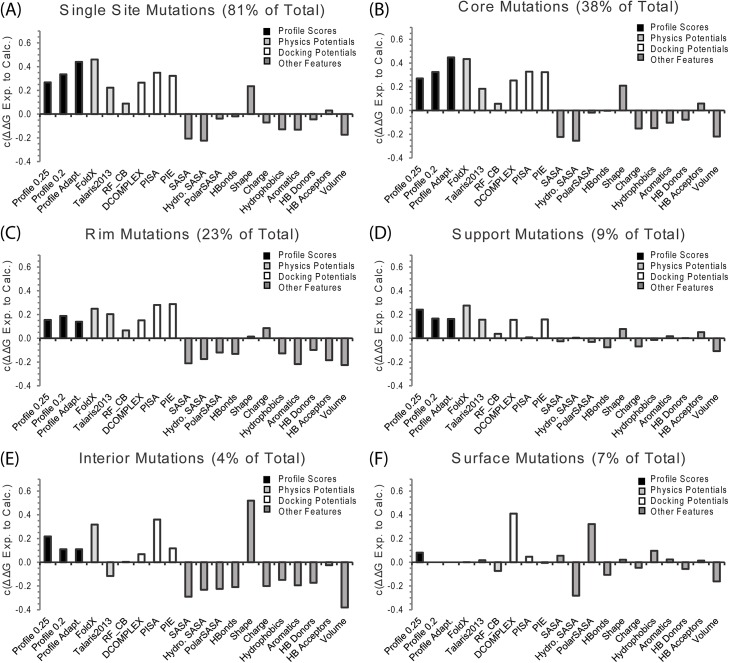
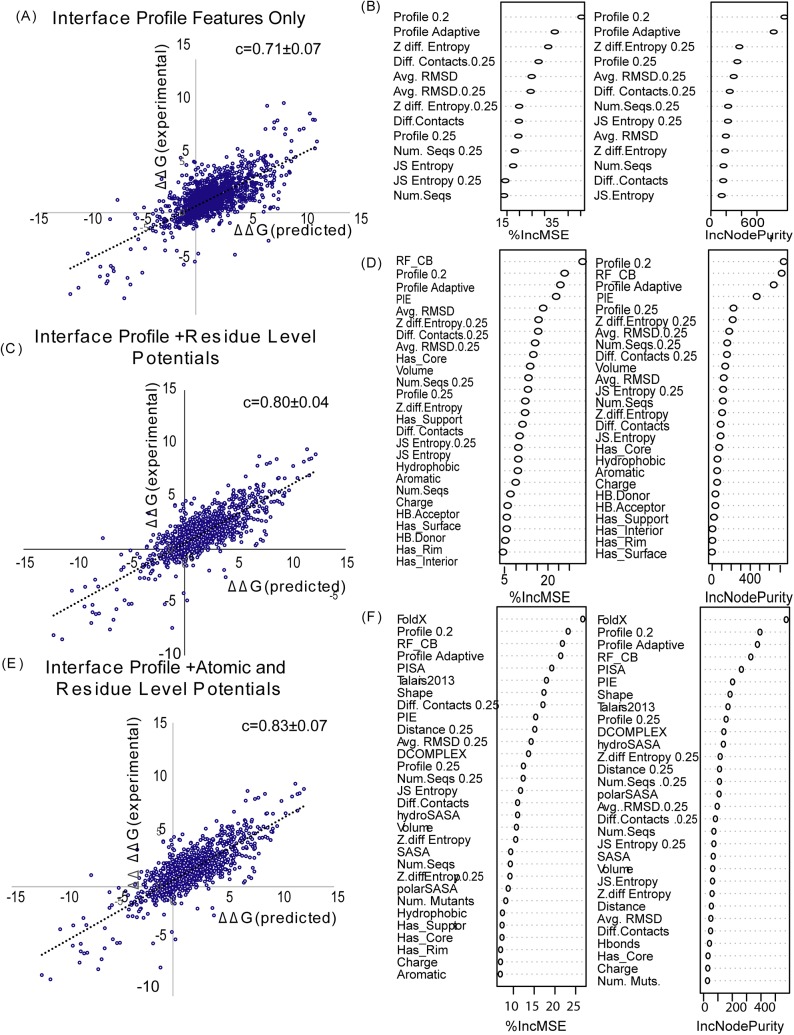
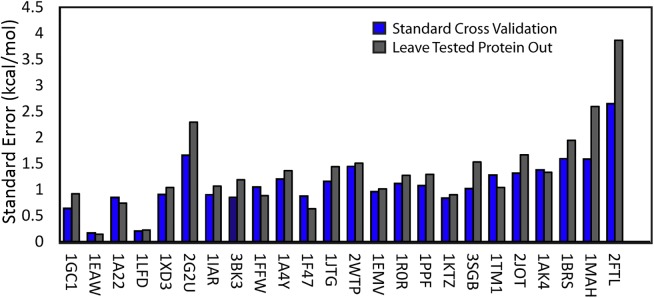
References
-
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nature genetics. 1999; 22(3): 231–8. - PubMed
-
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001; 409(6822): 928–33. - PubMed
-
- Kortemme T, Baker D. Computational design of protein-protein interactions. Current opinion in chemical biology. 2004; 8(1): 91–7. - PubMed
-
- Leavitt S, Freire E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Current opinion in structural biology. 2001; 11(5): 560–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
