

Genetic testing in steroid-resistant nephrotic syndrome: when and how?
- PMID: 26507970
- PMCID: PMC6367944
- DOI: 10.1093/ndt/gfv355
Genetic testing in steroid-resistant nephrotic syndrome: when and how?
Abstract
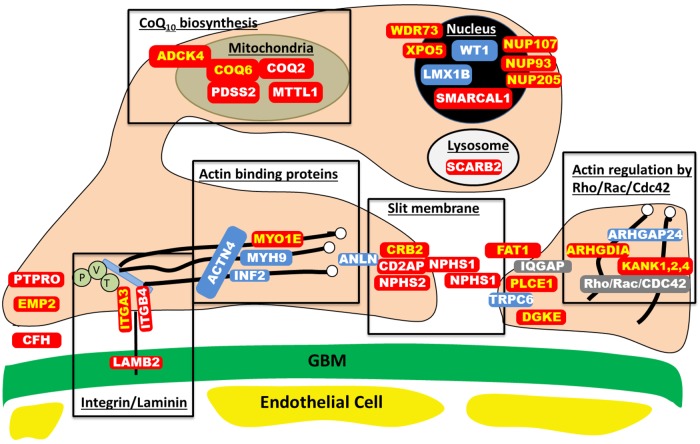
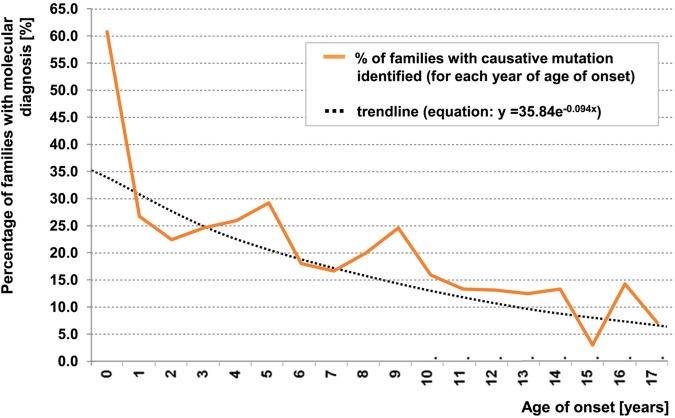
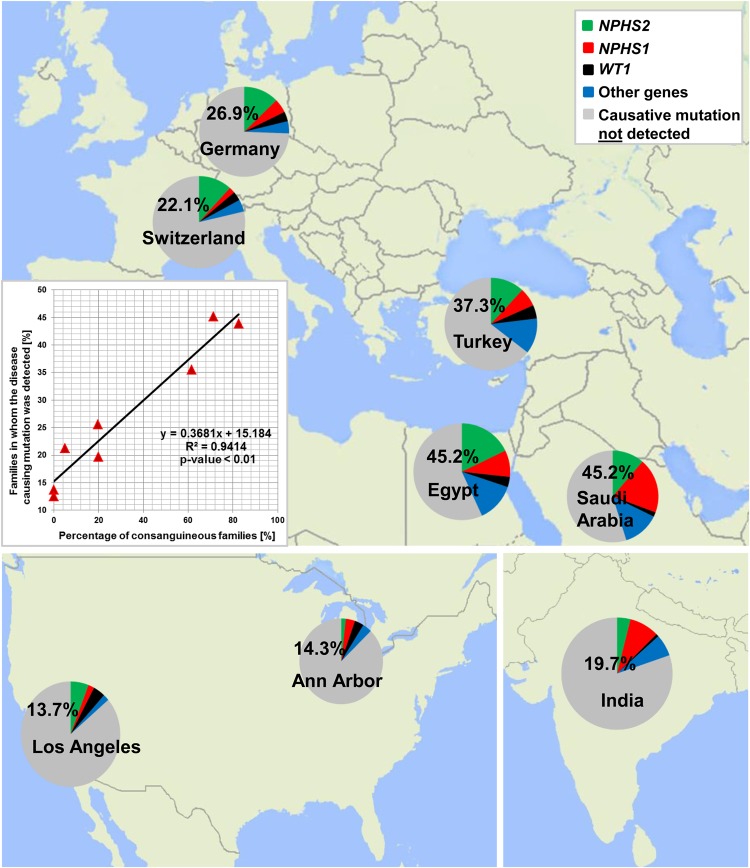
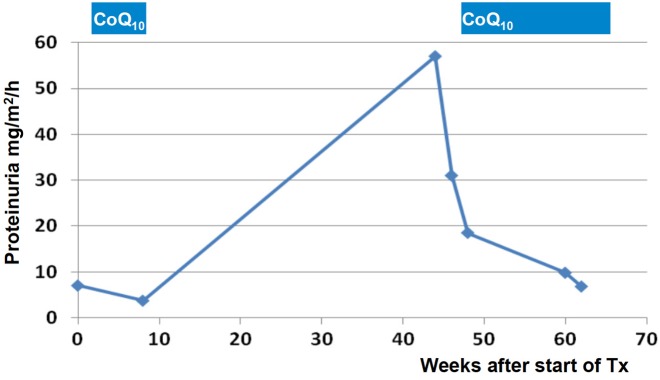
Steroid-resistant nephrotic syndrome (SRNS) represents the second most frequent cause of chronic kidney disease in the first three decades of life. It manifests histologically as focal segmental glomerulosclerosis (FSGS) and carries a 33% risk of relapse in a renal transplant. No efficient treatment exists. Identification of single-gene (monogenic) causes of SRNS has moved the glomerular epithelial cell (podocyte) to the center of its pathogenesis. Recently, mutations in >30 recessive or dominant genes were identified as causing monogenic forms of SRNS, thereby revealing the encoded proteins as essential for glomerular function. These findings helped define protein interaction complexes and functional pathways that could be targeted for treatment of SRNS. Very recently, it was discovered that in the surprisingly high fraction of ∼30% of all individuals who manifest with SRNS before 25 years of age, a causative mutation can be detected in one of the ∼30 different SRNS-causing genes. These findings revealed that SRNS and FSGS are not single disease entities but rather are part of a spectrum of distinct diseases with an identifiable genetic etiology. Mutation analysis should be offered to all individuals who manifest with SRNS before the age of 25 years, because (i) it will provide the patient and families with an unequivocal cause-based diagnosis, (ii) it may uncover a form of SRNS that is amenable to treatment (e.g. coenzyme Q10), (iii) it may allow avoidance of a renal biopsy procedure, (iv) it will further unravel the puzzle of pathogenic pathways of SRNS and (v) it will permit personalized treatment options for SRNS, based on genetic causation in way of 'precision medicine'.
Keywords: clinical genetic testing; molecular genetics; monogenic disease; pathogenesis of nephrotic syndrome; steroid-resistant nephrotic syndrome (SRNS).
© The Author 2015. Published by Oxford University Press on behalf of ERA-EDTA. All rights reserved.
Figures
References
-
- Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 2007; 71: 1205–1214 - PubMed
-
- Smith JM, Stablein DM, Munoz R et al. . Contributions of the transplant registry: the 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). Pediatr Transplant 2007; 11: 366–373 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
