

Oxidative Stress in Dilated Cardiomyopathy Caused by MYBPC3 Mutation
- PMID: 26508994
- PMCID: PMC4609873
- DOI: 10.1155/2015/424751
Oxidative Stress in Dilated Cardiomyopathy Caused by MYBPC3 Mutation
Abstract
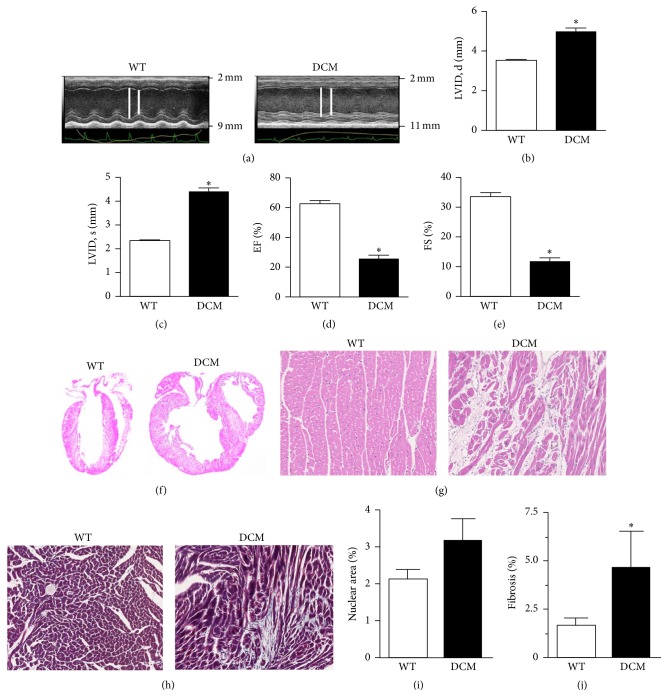
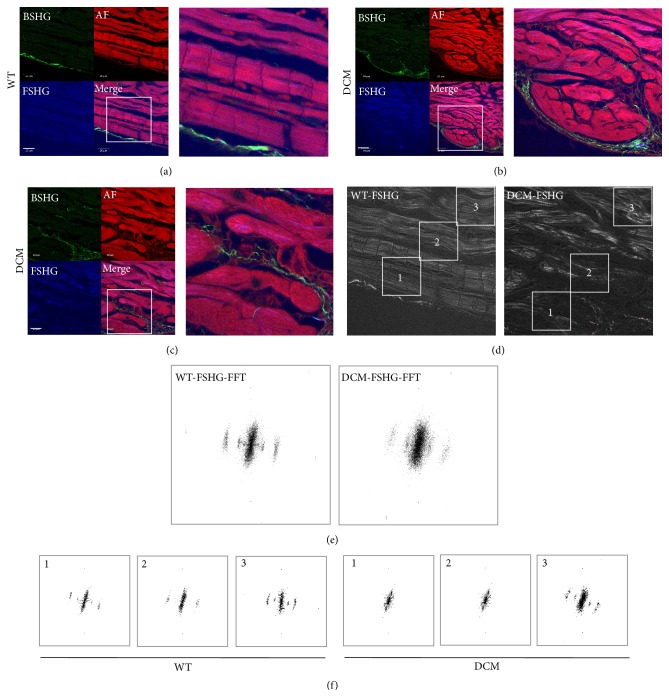
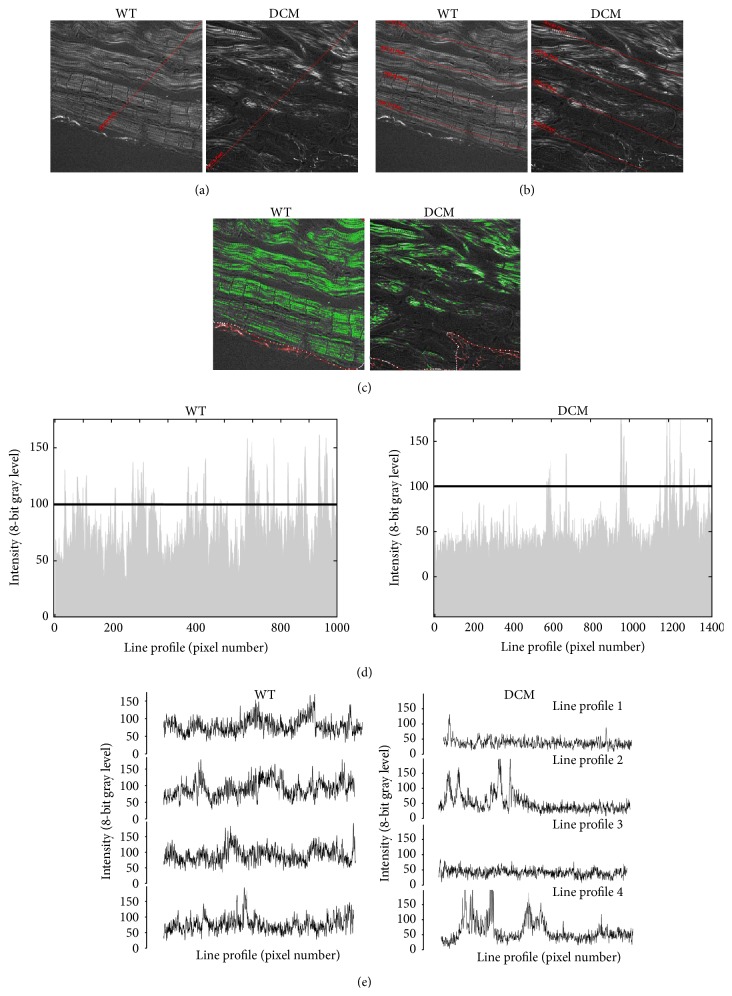
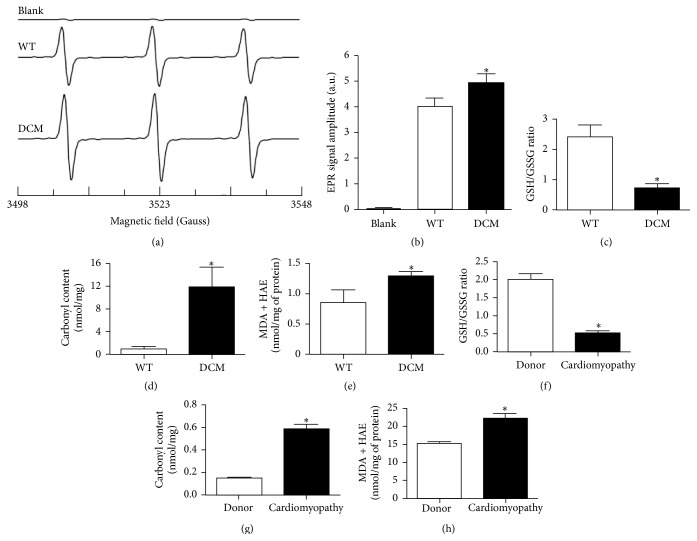
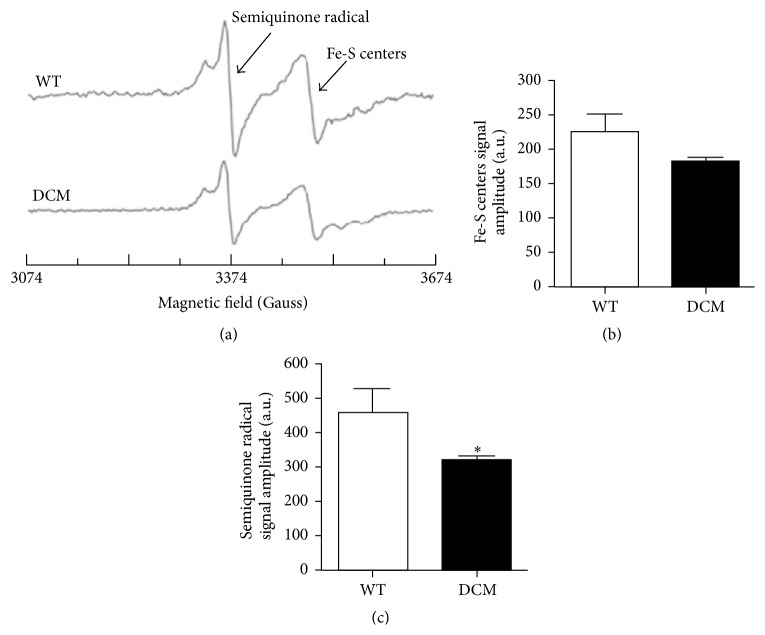
Cardiomyopathies can result from mutations in genes encoding sarcomere proteins including MYBPC3, which encodes cardiac myosin binding protein-C (cMyBP-C). However, whether oxidative stress is augmented due to contractile dysfunction and cardiomyocyte damage in MYBPC3-mutated cardiomyopathies has not been elucidated. To determine whether oxidative stress markers were elevated in MYBPC3-mutated cardiomyopathies, a previously characterized 3-month-old mouse model of dilated cardiomyopathy (DCM) expressing a homozygous MYBPC3 mutation (cMyBP-C((t/t))) was used, compared to wild-type (WT) mice. Echocardiography confirmed decreased percentage of fractional shortening in DCM versus WT hearts. Histopathological analysis indicated a significant increase in myocardial disarray and fibrosis while the second harmonic generation imaging revealed disorganized sarcomeric structure and myocyte damage in DCM hearts when compared to WT hearts. Intriguingly, DCM mouse heart homogenates had decreased glutathione (GSH/GSSG) ratio and increased protein carbonyl and lipid malondialdehyde content compared to WT heart homogenates, consistent with elevated oxidative stress. Importantly, a similar result was observed in human cardiomyopathy heart homogenate samples. These results were further supported by reduced signals for mitochondrial semiquinone radicals and Fe-S clusters in DCM mouse hearts measured using electron paramagnetic resonance spectroscopy. In conclusion, we demonstrate elevated oxidative stress in MYPBC3-mutated DCM mice, which may exacerbate the development of heart failure.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
