

Library preparation methodology can influence genomic and functional predictions in human microbiome research
- PMID: 26512100
- PMCID: PMC4653211
- DOI: 10.1073/pnas.1519288112
Library preparation methodology can influence genomic and functional predictions in human microbiome research
Abstract
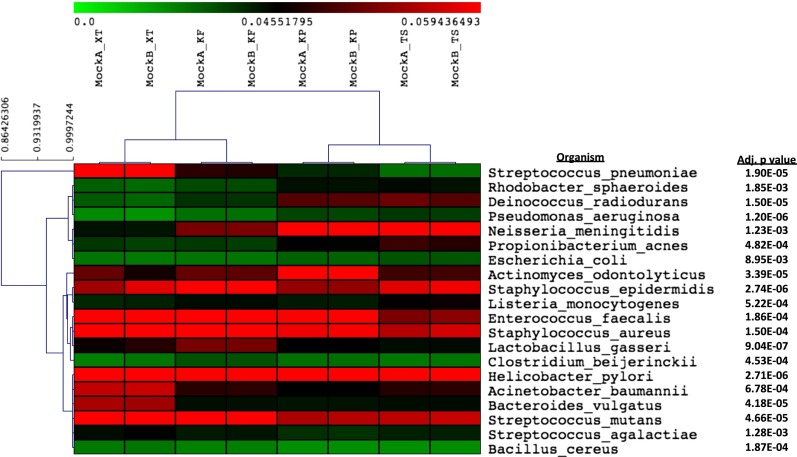
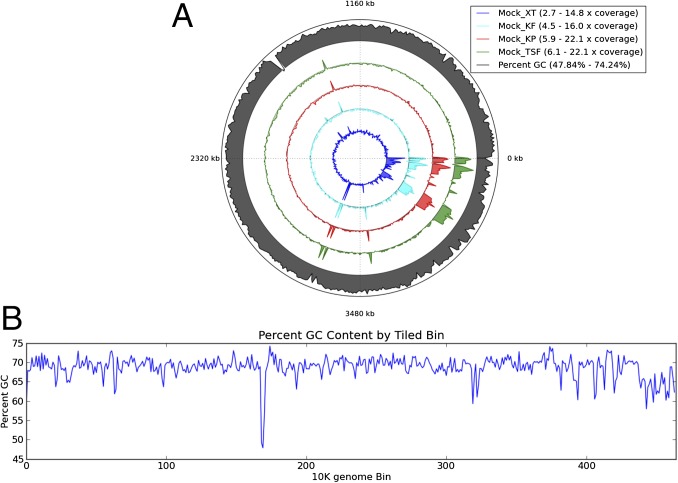
Observations from human microbiome studies are often conflicting or inconclusive. Many factors likely contribute to these issues including small cohort sizes, sample collection, and handling and processing differences. The field of microbiome research is moving from 16S rDNA gene sequencing to a more comprehensive genomic and functional representation through whole-genome sequencing (WGS) of complete communities. Here we performed quantitative and qualitative analyses comparing WGS metagenomic data from human stool specimens using the Illumina Nextera XT and Illumina TruSeq DNA PCR-free kits, and the KAPA Biosystems Hyper Prep PCR and PCR-free systems. Significant differences in taxonomy are observed among the four different next-generation sequencing library preparations using a DNA mock community and a cell control of known concentration. We also revealed biases in error profiles, duplication rates, and loss of reads representing organisms that have a high %G+C content that can significantly impact results. As with all methods, the use of benchmarking controls has revealed critical differences among methods that impact sequencing results and later would impact study interpretation. We recommend that the community adopt PCR-free-based approaches to reduce PCR bias that affects calculations of abundance and to improve assemblies for accurate taxonomic assignment. Furthermore, the inclusion of a known-input cell spike-in control provides accurate quantitation of organisms in clinical samples.
Keywords: genomics; microbiome; sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Kim M, Yu Z. Variations in 16S rRNA-based microbiome profiling between pyrosequencing runs and between pyrosequencing facilities. J Microbiol. 2014;52(5):355–365. - PubMed
-
- Schmidt TS, Matias Rodrigues JF, von Mering C. Limits to robustness and reproducibility in the demarcation of operational taxonomic units. Environ Microbiol. 2015;17(5):1689–1706. - PubMed
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
