

Gapped sequence alignment using artificial neural networks: application to the MHC class I system
- PMID: 26515819
- PMCID: PMC6402319
- DOI: 10.1093/bioinformatics/btv639
Gapped sequence alignment using artificial neural networks: application to the MHC class I system
Abstract
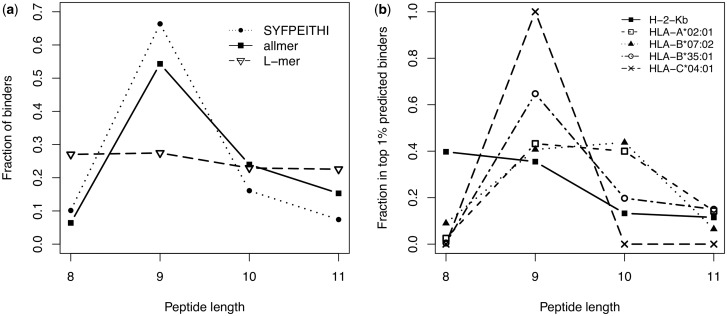
Motivation: Many biological processes are guided by receptor interactions with linear ligands of variable length. One such receptor is the MHC class I molecule. The length preferences vary depending on the MHC allele, but are generally limited to peptides of length 8-11 amino acids. On this relatively simple system, we developed a sequence alignment method based on artificial neural networks that allows insertions and deletions in the alignment.
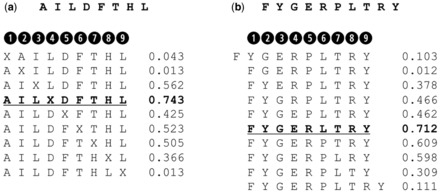
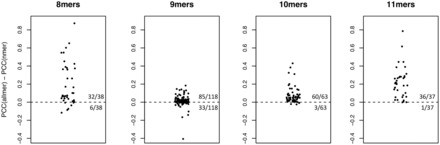
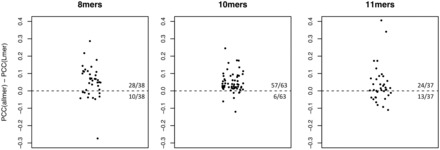
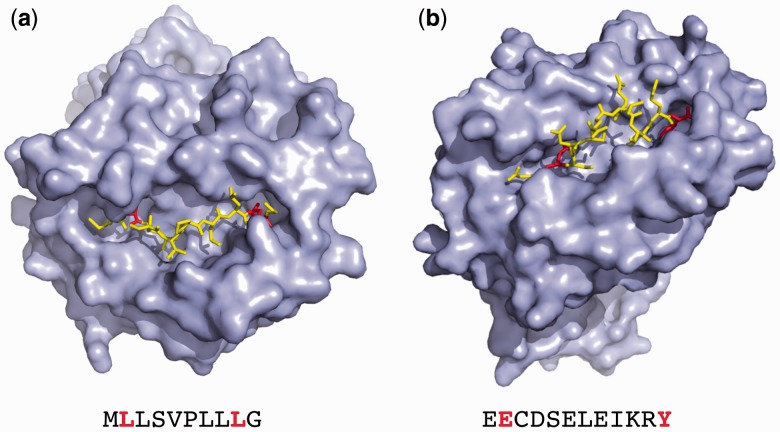
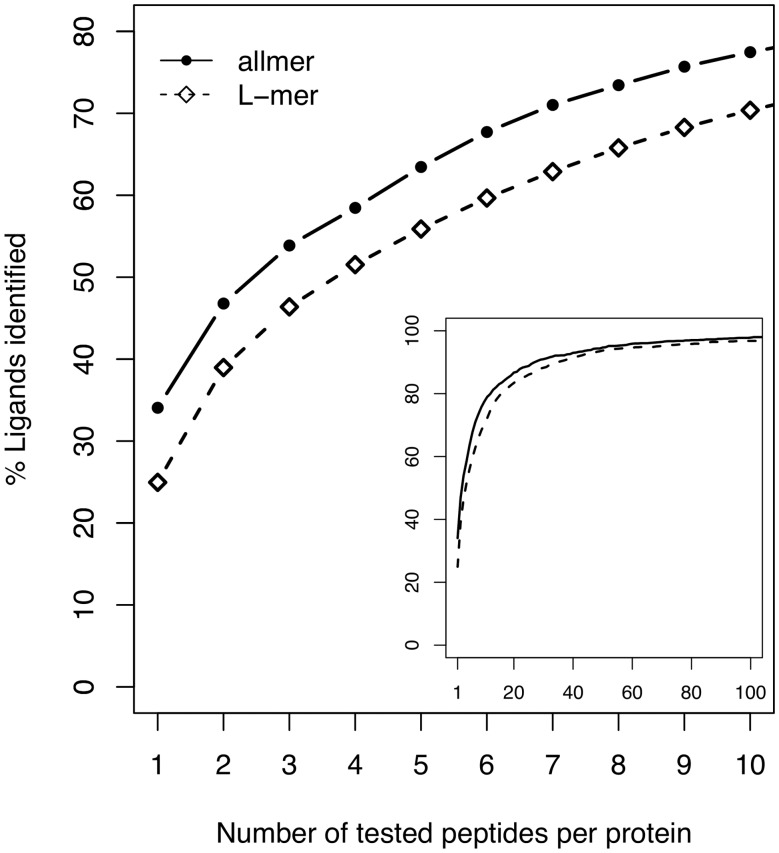
Results: We show that prediction methods based on alignments that include insertions and deletions have significantly higher performance than methods trained on peptides of single lengths. Also, we illustrate how the location of deletions can aid the interpretation of the modes of binding of the peptide-MHC, as in the case of long peptides bulging out of the MHC groove or protruding at either terminus. Finally, we demonstrate that the method can learn the length profile of different MHC molecules, and quantified the reduction of the experimental effort required to identify potential epitopes using our prediction algorithm.
Availability and implementation: The NetMHC-4.0 method for the prediction of peptide-MHC class I binding affinity using gapped sequence alignment is publicly available at: http://www.cbs.dtu.dk/services/NetMHC-4.0.
© The Author 2015. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Burrows S.R., et al. (2006) Have we cut ourselves too short in mapping CTL epitopes?. Trends Immunol., 27, 11–16. - PubMed
-
- Collins E.J., et al. (1994) Three-dimensional structure of a peptide extending from one end of a class I MHC binding site. Nature, 371, 626–629. - PubMed
-
- Deres K., et al. (1992) Preferred size of peptides that bind to H-2 Kb is sequence dependent. Eur. J. Immunol., 22, 1603–1608. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
