

Tumor cell survival pathways activated by photodynamic therapy: a molecular basis for pharmacological inhibition strategies
- PMID: 26516076
- PMCID: PMC4661210
- DOI: 10.1007/s10555-015-9588-7
Tumor cell survival pathways activated by photodynamic therapy: a molecular basis for pharmacological inhibition strategies
Abstract
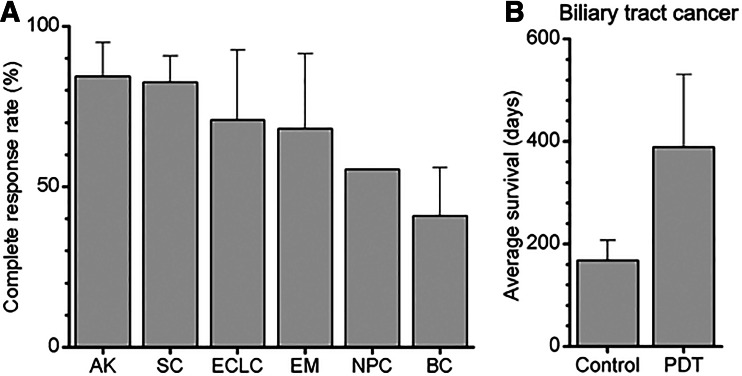
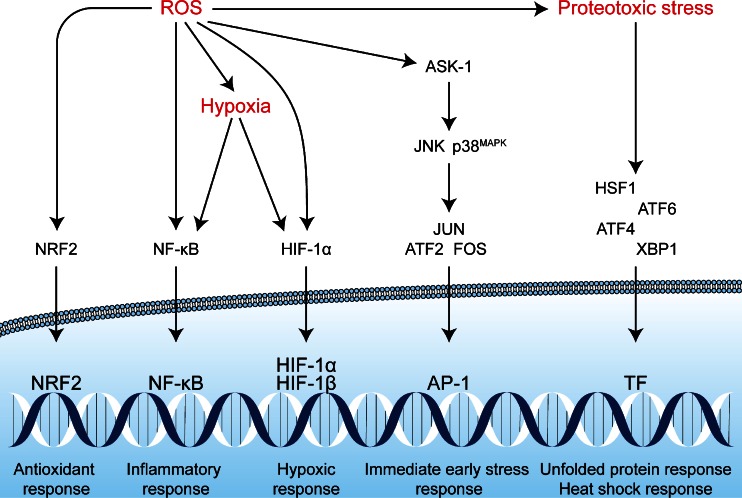
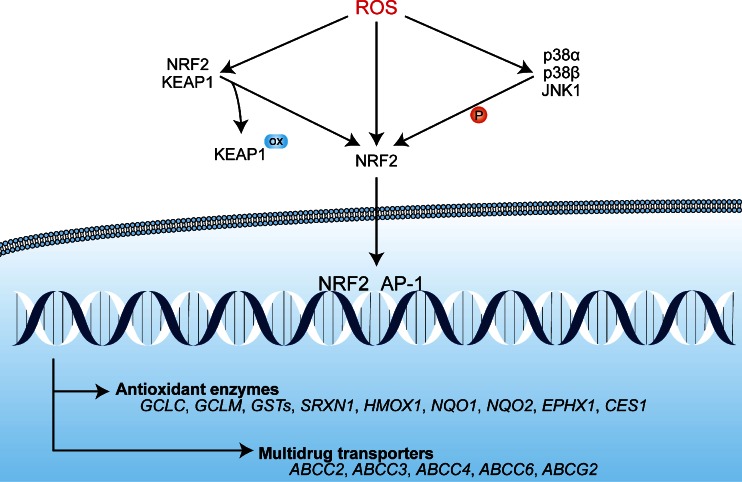
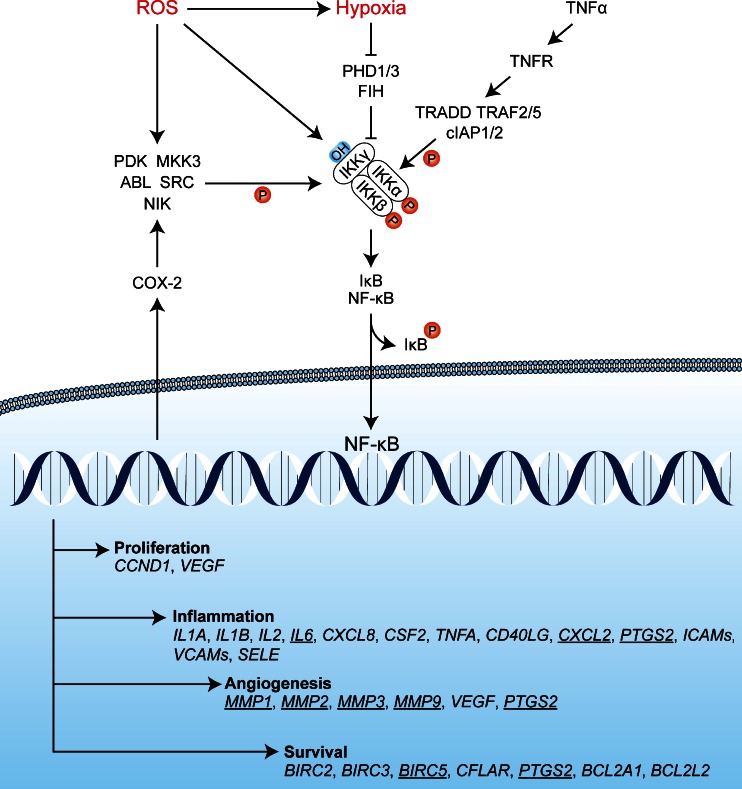
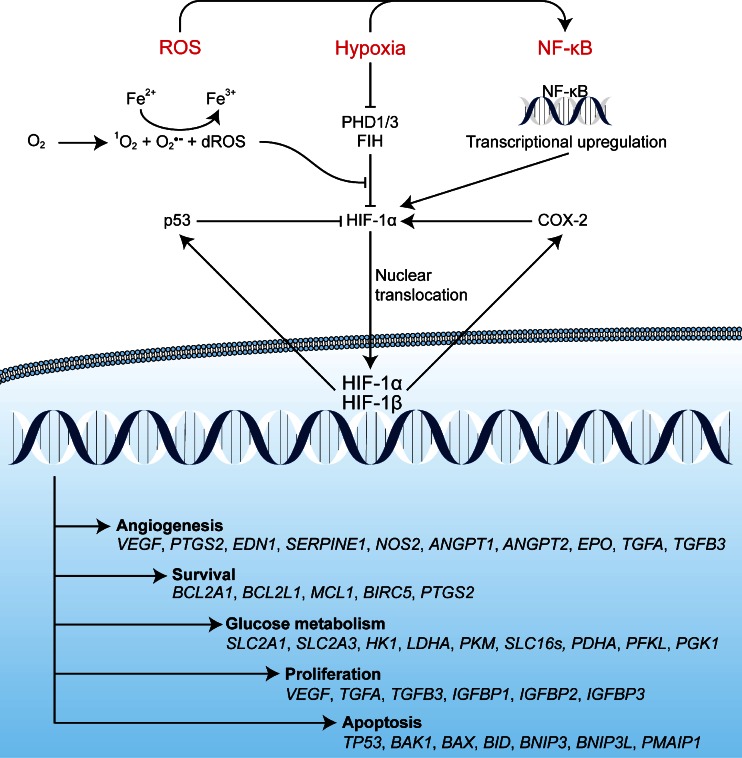
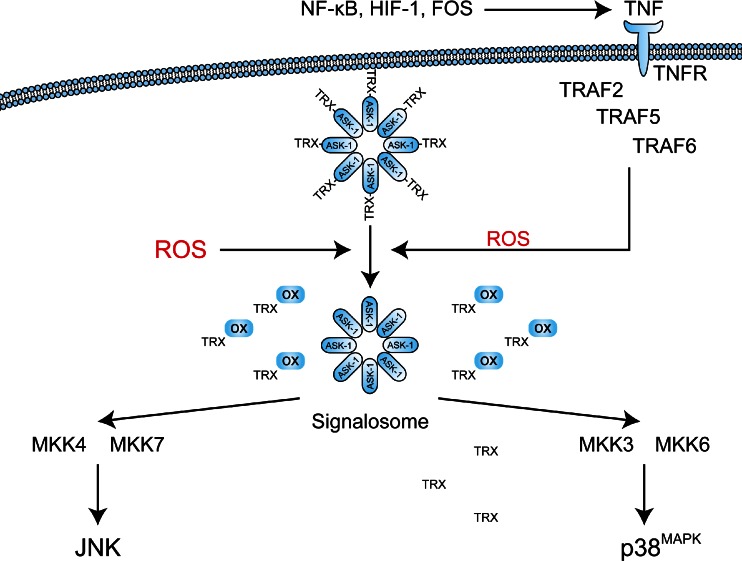
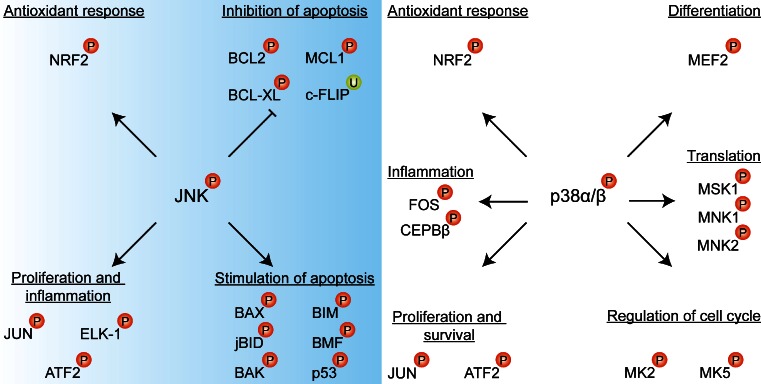
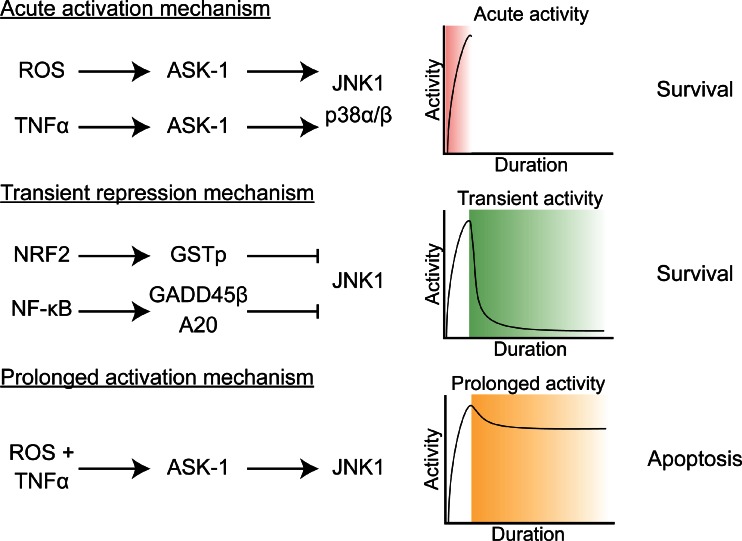
Photodynamic therapy (PDT) has emerged as a promising alternative to conventional cancer therapies such as surgery, chemotherapy, and radiotherapy. PDT comprises the administration of a photosensitizer, its accumulation in tumor tissue, and subsequent irradiation of the photosensitizer-loaded tumor, leading to the localized photoproduction of reactive oxygen species (ROS). The resulting oxidative damage ultimately culminates in tumor cell death, vascular shutdown, induction of an antitumor immune response, and the consequent destruction of the tumor. However, the ROS produced by PDT also triggers a stress response that, as part of a cell survival mechanism, helps cancer cells to cope with the PDT-induced oxidative stress and cell damage. These survival pathways are mediated by the transcription factors activator protein 1 (AP-1), nuclear factor E2-related factor 2 (NRF2), hypoxia-inducible factor 1 (HIF-1), nuclear factor κB (NF-κB), and those that mediate the proteotoxic stress response. The survival pathways are believed to render some types of cancer recalcitrant to PDT and alter the tumor microenvironment in favor of tumor survival. In this review, the molecular mechanisms are elucidated that occur post-PDT to mediate cancer cell survival, on the basis of which pharmacological interventions are proposed. Specifically, pharmaceutical inhibitors of the molecular regulators of each survival pathway are addressed. The ultimate aim is to facilitate the development of adjuvant intervention strategies to improve PDT efficacy in recalcitrant solid tumors.
Keywords: Antioxidant response; Apoptosis signaling kinase 1; ER stress; Heat shock factor 1; Inflammatory response; Proteotoxic stress.
Figures
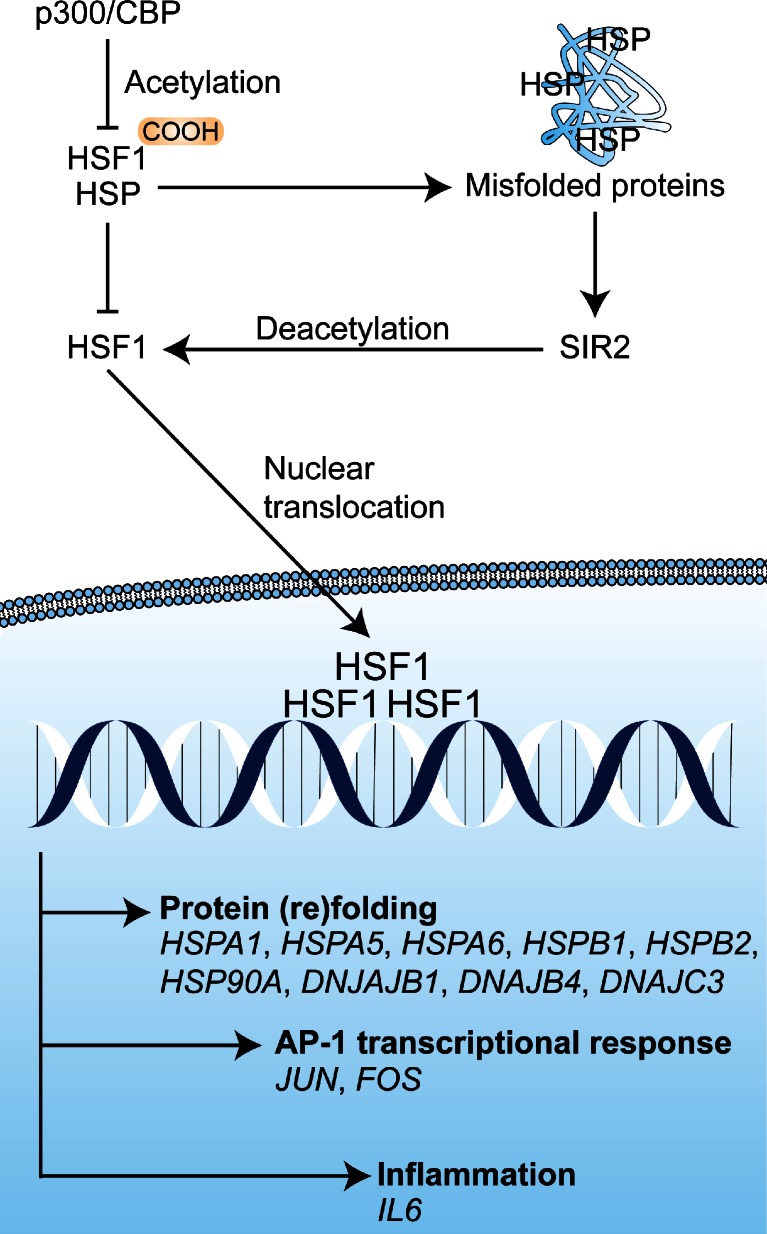
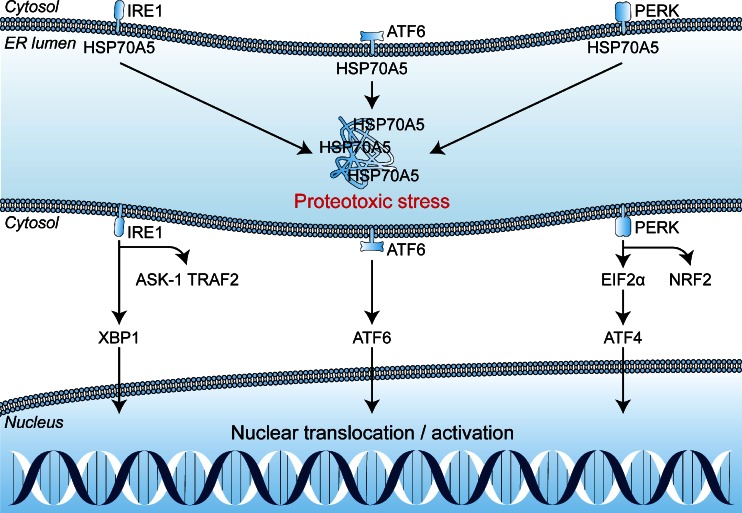
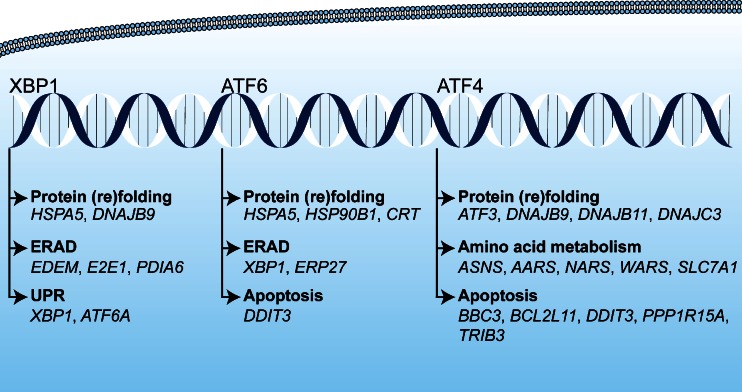
References
-
- Kübler A, Niziol C, Sidhu M, Dünne A, Werner JA. Analysis of cost effectiveness of photodynamic therapy with Foscan (Foscan PDT) in comparison to palliative chemotherapy in patients with advanced head-and-neck tumors in Germany. Laryngo- Rhino- Otologie. 2005;84:725–732. doi: 10.1055/s-2005-861048. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
